参考书籍
《医学遗传学》第7版,左伋等主编。北京人民卫生出版社出版,2018,全国高等学校五年制本科临床医学专业第九轮规划教材,ISBN 978-7-117-26440-2。
基于疾病的遗传学数据分析
遗传数据库
遗传数据库(genetic database)是储存在计算机内的、有组织的、可共享的遗传数据(genetic data)的集合。
国际上最权威、最主要的三大核酸序列数据库,包括
- 美国国家生物技术信息中心(National Center for Biotechnology Information,
NCBI)维护的GenBank; - 欧洲分子生物学研究室(European Molecular Biology Laboratory,
EMBL)下属的欧洲生物信息学研究所(European Bioinformatics Institute,EBI)维护的EMBL-EBI; - 日本国立遗传学研究所维护的
DDBJ(DNA Data Bank of Japan)
是目前最具有影响力的生命科学数据库。
常用的人类基因组与遗传数据库
| 序号 | 数据库(或网站) | 网址 |
|---|---|---|
| 1 | OMIM:权威的遗传病分子医学百科全书和数据库 | www.omim.org |
| 2 | GeneTests:权威、常用的遗传病基因检测和基因诊断数据库 | www.genetests.org |
| 3 | HGMD:权威、完整的人类遗传病或与人类遗传病相关的核基因突变数据库 | www.hgmd.cf.ac.uk |
| 4 | GeneCards:信息量非常完整的人类基因综合数据库 | www.genecards.org |
| 5 | Ensembl:有关人类基因组和其他物种基因组的全面资源的综合性基因组数据库。由欧洲生物信息学会和Wellcome 基金会Sanger研究所共同维护 | www.ensembl.org |
| 6 | HGP相关资料数据库 | www.genome.gov/10001772 http://genome.ucsc.edu/cgi-bin/hgGateway www.ensembl.org/Homo_sapiens/Info/Index |
| 7 | dbSNP和dbVar:NCBI旗下的SNP数据库和结构变异数据库,收录包括单核苷酸变异、微卫星DNA、indel和CNV等各种变异体 | www.ncbi.nlm.nih.gov/snp/ www.ncbi.nlm.nih.gov/dbvar/ |
| 8 | ClinVar:NCBI创建于2013年的与疾病相关的遗传变异数据库。内含超过十几万份独特突变的临床注释 | www.ncbi.nlm.nih.gov/clinvar |
| 9 | G1K计划(国际千人基因组计划):对所有公众开放的包含几千个个体的基因组数据的数据库 | www.internationalgenome.org |
| 10 | Database of Genomic Variants(基因组变异体数据库):收录人基因组的结构变异体。截至2012年的统计,总条目超过400 000个,其中CNV超过200 000种,倒位超过1000种,indel超过34 000种 | http://dgv.tcag.ca |
| 11 | JSNP:日本SNP数据库。由日本东京大学维护 | http://snp.ims.u-tokyo.ac.jp/ |
| 12 | UCSC基因组生物信息学数据库:美国加州大学Santa Cruz分校创建和维护的权威的基因组浏览数据库 | http://genome.ucsc.edu/ |
| 13 | HUGO(Human Genome Organization,人类基因组协会) | www.hugo-international.org |
| 14 | HapMap计划:提供数百万个SNP,标志着人类基因组的研究"从一个个体的基因组参考序列到人类基因组的多样性"的重要里程碑 | www.hapmap.org ftp://ftp.ncbi.nlm.nih.gov/hapmap/ |
| 15 | G10K计划(英国万人基因组计划) | www.genomicsengland.co.uk/the-100000-genomes-project/ |
| 16 | ExAC:已收录超过6万例个体外显子组测序结果的数据库。利用ExAC数据库可以避免遗传性误诊 | http://exac.broadinstitute.org/ |
| 17 | EVS:已收录超过5000例个体的外显子组测序结果的数据库,旨在揭示心脏病、呼吸系统疾病、血液病的分子病因,从而进行精准诊治 | http://evs.gs.washington.edu/EVS/ |
| 18 | BROAD研究所:提供人类基因定位、测序、各种分析软件的信息 | www.broad.mit.edu |
| 19 | 预测基因变异体是否具有临床致病性的软件 VEP SIFT POLYPHEN2 ALIGNGVGD |
www.ensembl.org/info/docs/tools/vep/ http://sift.jcvi.org/ http://genetics.bwh.harvard.edu/pph2/ http://agvgd.hci.utah.edu/agvgd_input.php |
| 20 | Decipher:"Database of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources"的缩写,收录了超过200家研究中心上传的10 000多例案例的信息,主要用于筛查比对CNV | http://decipher.sanger.ac.uk/ |
| 21 | MitoMap:最权威的人类线粒体参考基因序列库、相关基因突变与疾病数据库 | www.mitomap.org |
| 22 | London Medical Database Online(在线伦敦医疗数据库):与Face2Gene公司合办,包含Winter-Baraitser畸形学数据库、Baraitser-Winter神经遗传学数据库和伦敦眼科遗传学数据库 | www.fdna.com/london-medical-databases-online |
| 23 | GTR(Genetic Testing Registry):NCBI收录有关遗传病基因检测信息数据库 | www.ncbi.nlm.nih.gov/gtr |
| 24 | GHR(Genetics Home Reference):NIH旗下的美国国立医学图书馆(National Library of Medicine,NLM)创建和维护的医学遗传学网站 | http://ghr.nlm.nih.gov/ |
| 25 | PharmGKB:药物遗传学和药物基因组学的权威数据库 | www.pharmgkb.org |
| 26 | ClinicalTrials.gov:NIH旗下的NLM维护的关于疾病的临床试验信息的数据库。截至2017年9月25日,收录了美国50个州和全球200个国家的255 652个临床试验研究资料 | http://clinicaltrials.gov/ |
| 27 | The Journal of Gene Medicine Clinical Trial:收录了全球关于基因治疗的临床试验研究信息 | www.wiley.com/legacy/wileychi/genmed/clinical/ |
| 28 | ELSI:人类基因组学的伦理、法律、社会问题/影响 | www.nhgri.nih.gov/PolicyEthics/ |
| 29 | Orphanet:提供罕见病和罕见病药物信息的开放门户网站,目的在于提高罕见病的诊断、护理和治疗效果 | www.orpha.net |
| 30 | Genetic Alliance UK:英国一家为罕患各种遗传病的患者及其亲属提供相关帮助的公益机构 | www.geneticalliance.org.uk |
| 31 | NORD:即"National Organization for Rare Disorders"的缩写,美国全国罕见病组织联合体。数据库中收录了1200多种遗传病 | http://rarediseases.org/ |
注:HCP = human genome project(人类基因组计划);CNV = copy number variant(拷贝数变异体);SNP = single nucleotide polymorphism(单核苷酸多态性)
常用遗传数据库介绍
OMIM
OMIM包括所有已知的遗传病、遗传决定的性状及其基因,除了简略描述各种疾病的临床特征、诊断、鉴别诊断、治疗与预防外,还提供已知有关致病基因的连锁关系、染色体定位、基因的组成结构和功能、表型-基因型相关性、表型的系列信息、国际疾病分类号、动物模型等资料,并附有经过缜密筛选的相关参考文献。
OMIM 制定的各种遗传病、性状、基因的编号,简称 OMIM 编号,其 6 位数字,为全世界所公认。有关疾病的报道必须冠以 OMIM 编号,以明确所讨论的是哪一种遗传病。因此,OMIM 是研究疾病与基因相关性的重要依据,为每一位医务工作者所必须掌握的核心资源。
例如,呈常染色体显性遗传的表皮松解性掌跖角化症,其 OMIM 编号为"144200"。
OMIM 编号中的有关含义如下:
-
编号前有"
*":表示该条目(entry)是一个基因。 -
编号前有"
#":表示该条目是一个描述性的条目,通常为一种表型(疾病或性状),而非一个特定的基因座。 -
编号前有"
+":表示该条目包含了已知 DNA 序列的基因以及表型的相关描述。 -
编号前有"
%":表示该条目描述了一种已经明确了的孟德尔表型或一个未知分子基础的表型基因座。 -
编号前无任何符号:表示该条目尽管被怀疑为孟德尔性状,但是否为孟德尔遗传方式的表型信息尚未明确,或尚不能将该条目列为一个单独的条目。
-
编号前有"
^":表示该条目现已不存在,或已从 OMIM 数据库中移除,或已被合并至其他条目中。 -
编号为"
100000~199999,200000~299999"的条目:表示为常染色体基因座或表型(创建于 1994年 5 月 15 日之前的相关条目)。 -
编号为"
300000~399999"的条目:表示为 X 染色体连锁基因座或表型。 -
编号为"
400000~499999"的条目:表示为 Y 染色体连锁基因座或表型。 -
编号为"
500000~599999"的条目:表示为线粒体遗传基因座或表型。 -
编号为"
600000~699999"的条目:表示为常染色体基因座或表型(创建于 1994 年 5 月 15 日之后的相关条目)。
GeneTests
GeneTests(www.genetests.org)是最权威、最常用的有关遗传病基因检测和基因诊断的数据库。
GeneTests 囊括了全球经标准化资质审核之后准许进行基因检测和基因诊断的所有遗传病、基因、医院、独立医学检验所、高校实验室、基因诊断公司等信息。
GeneTests 数据库另一大亮点是其"GeneReviews(基因综述)"栏目。GeneReviews 是由一位或几位相关遗传病领域的专家撰写,并经同行审校的综述性论文,提供所描述的遗传病的概况、致病基因信息、突变检测手段、疾病的诊治方法和遗传咨询等应用的最新且权威的信息。
GeneReviews 目前包含 694 篇综述,约 95% 的综述瞄准单基因病或基因,另有约 5% 的综述探讨的是耳聋、Alzheimer病等常见病。GeneReviews 每月增加数种,每篇综述 1~2 年内更新一次,并根据当今的遗传病研究进展随时进行修订。
在 NCBI 网站上,GeneReviews 有专门的免费链接地址:
www.ncbi.nlm.nih.gov/books/NBK1116/
HGMD
HGMD(Human Gene Mutation Database)(www.hgmd.cf.ac.uk)即"人类基因突变数据库",是由英国威尔士 Cardiff 大学医学遗传学研究所创建和维护的著名通用型数据库,全面收录了导致人类遗传病或与人类遗传病相关的核基因突变。
HGMD 建立的初衷是用于基因突变机制的分析,但由于其收录最新的、完整的有关人类疾病基因突变谱的参考数据,包括单碱基置换(如基因编码序列中的错义突变、无义突变以及 DNA 调控和剪接区域中的点突变)、微小缺失(micro-deletion)、微小插入(micro-insertion)、插缺(indel)、重复序列扩增、大的基因损伤(大片段缺失、大片段插入和基因扩增)、复杂性基因重组等,因而具有很高的权威性声誉,一直为学者们广泛应用。
HGMD 分为免费公共版和专业版两种。
免费公共版提供给注册学术用户和非盈利用户的只是那些已收录了 3 年以上的数据,而更完整的、每 3 个月更新一次的专业版数据需要通过付费形式,从 HGMD 的商业合作伙伴 BIOBASE GmbH公司那里获得。
显然,HGMD 专业版集实时更新、结果下载、高级检索等多项功能于一体:
- 包含超过 179 000 份基因突变报告,涵盖突变所在的基因组位置、序列的详细信息,并可链接到参考文献及其他公共平台,如
dbSNP、OMIM等; - 包含超过 6800 份总结报告,对给定基因的 6 种不同的致病变异进行描述,并罗列所有已知的遗传病的突变;
- 高级搜索功能:可基于核酸/氨基酸的改变,或在特异序列、剪接位点、调控区域中的位置进行精确的突变检索;
- 可视化的突变展示工具:为所比对的 DNA 序列或蛋白质序列提供图表描述,以不同的颜色区分突变核酸位点;
- 可在第三方基因组分析工具中导出自定义的突变文件。
因此,专业版可为从事基因组学、蛋白质组学、生物序列、人类疾病、基因突变等生物信息学研究的科研人员和医务工作者提供更便捷、全面的数据支持服务。
基因突变
基因突变的本质及其特性
发生在体细胞中的基因突变,即体细胞突变,虽然不会传递给后代个体,但是却能够通过突变细胞的分裂增殖而在后代子细胞中进行传递,形成突变的细胞克隆,成为具有体细胞遗传学特征的肿瘤病变甚或癌变的细胞组织病理学基础。
基因是具有特定遗传效应的DNA序列片段。 因此,无论是发生在生殖细胞中的基因突变,还是发生于体细胞中的基因突变,究其本质,实际上就是构成基因的DNA碱基组成与序列结构所发生的可遗传的变异,所以也具有一定的共同特性。
多向性
任何基因座上的基因,都有可能独立地发生多次不同的突变而形成其新的等位基因,这就是基因突变的多向性。
重复性
重复性是指已经发生突变的基因,在一定的条件下,还可能再次独立地发生突变而形成其另外一种新的等位基因形式。
亦即,对于任何一个基因来说,其突变并非仅囿于某一次或某几次的发生,而是会以一定的频率反复发生。
随机性
基因突变不仅是生物界普遍存在的一种遗传事件,而且,对于任何一种生物,任何一个个体,任何一个细胞乃至任何一个基因来说,突变的发生也都是随机的。 只是不同的物种、不同的个体、不同的细胞或者基因,其各自发生基因突变的频率可能并不完全相同而已。
可逆性
基因的突变是可逆的。 任何一种野生型基因都能够通过突变而形成其等位的突变型基因;反过来,突变型基因,也可以突变为其相应的野生型基因。 前者为正向突变,后者为回复突变。一般情况下,正向突变率总是远远高于回复突变率。
有害性
生物遗传性状的形成,是在长期的进化过程中与其赖以生存的自然环境相互作用、相互适应的结果,是自然选择的产物。而对这些性状具有决定性意义的基因一旦发生突变,通常都会对生物的生存带来消极或不利的影响,即有害性。
生殖细胞或受精卵中基因的突变是绝大多数人类遗传病发生的根本原因;体细胞突变则常常是肿瘤发生的病理遗传学基础。 然而,基因突变的有害性往往只是相对的,有条件的,也并非所有的基因突变都会对生物的生存及其种群繁衍带来不利或者有害的影响。
事实上,有些突变,往往只引起非功能性DNA序列组成的改变,却并不造成核酸和蛋白质正常功能的损害。
基因突变的形式
一般可归纳为静态突变和动态突变两种主要形式。
静态突变
静态突变(static mutation)是生物各世代中基因突变总是以一定的频率发生,并且能够使之随着世代的繁衍、交替而得以相对稳定地传递。
点突变
点突变(point mutation)是 DNA 多核苷酸链中单个碱基或碱基对的改变。
碱基替换
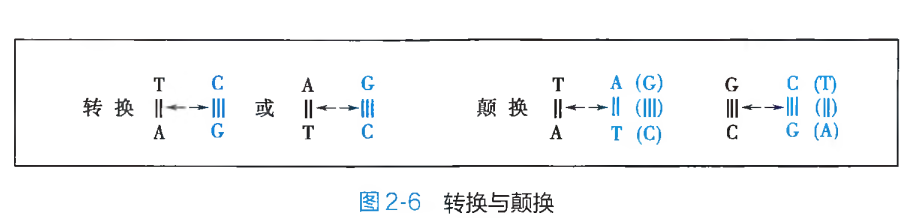
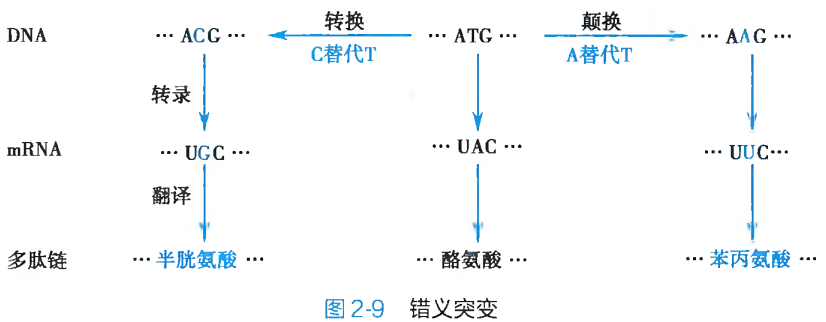
碱基替换(base substitution)是 DNA 分子多核苷酸链中原有的某一特定碱基或碱基对被其他碱基或碱基对置换、替代的突变形式。其具体表现为同类碱基或碱基对之间的替换及不同类碱基或碱基对之间的相互替换。同类碱基之间的替换,又被称之为转换(transition),即一种嘌呤碱或相应的嘧啶-嘧啶碱基对被另外一种嘌呤碱或相应的嘌呤-嘧啶碱基对所取代;如果某种嘌呤碱或其相应的嘧啶-嘧啶碱基对被另外一种嘧啶碱或其相应的嘧啶-嘌呤碱基对所置换,则称之为颠换(transvertion)(图 2-6)。
碱基替换只是原有碱基性质的改变,而并不涉及碱基数目的变化与异常。这种突变会因其作用对象的不同而产生不同的遗传学效应。如果被替换的是构成特定三联密码子单位的碱基或碱基对,则会造成:
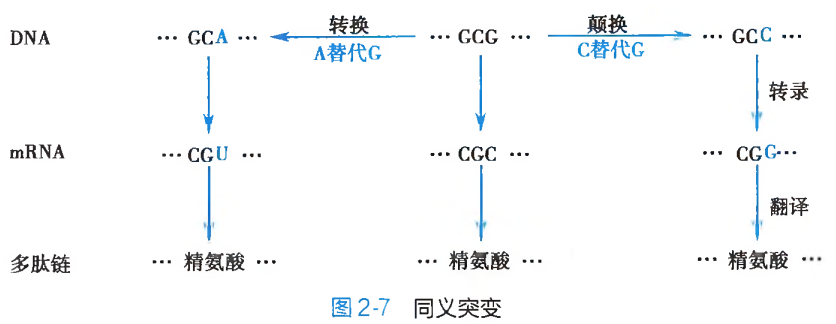
同义突变:由于存在遗传密码子的兼并现象,因此,替换的发生,尽管改变了原有三联遗传密码子的碱基组成,但是新、旧密码子所编码的氨基酸种类却依然保持不变。亦即新、旧密码子具有完全相同的编码意义(图 2-7),此为同义突变(same sense mutation)。同义突变并不产生相应的遗传表型突变效应。
无义突变:由于碱基替换而使得编码某一种氨基酸的三联体遗传密码子,变成为不编码任何氨基酸的终止密码 UAA、UAG 或 UGA 的突变形式被称之为无义突变(non-sense mutation)。此种突变,会引起翻译时多肽链合成延伸的提前终止(图 2-8A),造成多肽链的组成结构残缺及蛋白质功能的异常或丧失,最终会体现为导致遗传表型改变的致病效应。
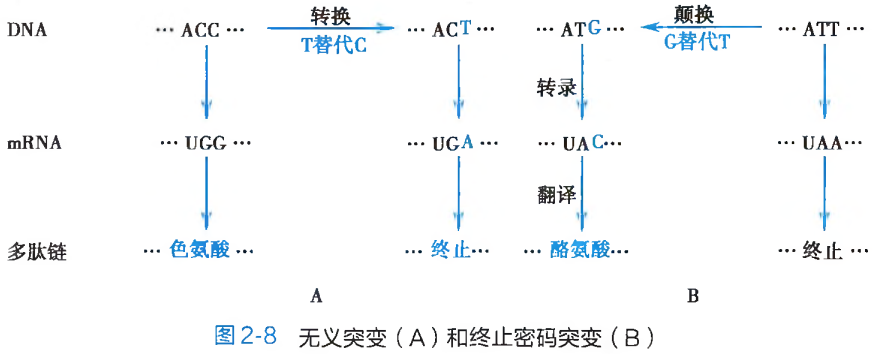
终止密码突变:如果因为碱基替换的发生,而使得 DNA 分子中某一终止密码变成了具有氨基酸编码功能的遗传密码子,此种突变形式即为终止密码突变(terminator codon mutation)(图 2-8B)。与无义突变相反,终止密码突变造成的将会使本应终止延伸的多肽链合成,非正常地持续进行。其结果也必然形成功能异常的蛋白质结构分子。错义突变:这是编码某种氨基酸的密码子经碱基替换后变成了另外一种氨基酸的密码子,从而在翻译时改变了多肽链中氨基酸的组成种类(图 2-9)。错义突变(missense mutation)的结果,必然地导致蛋白质多肽链原有功能的异常或丧失。人类的许多分子病和代谢病,就是因此而造成的。
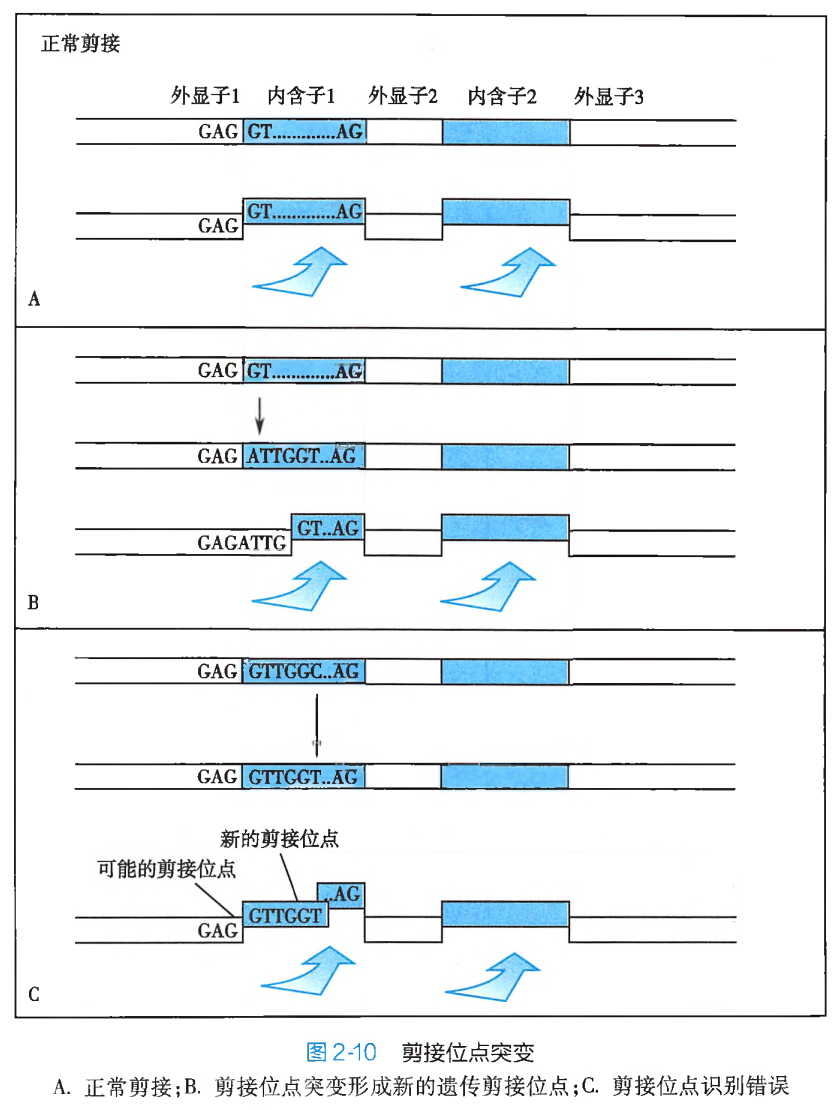
此外,碱基替换如果发生在 DNA 分子的非密码子组成结构区域,引起的将可能是调控序列或内含子与外显子剪接位点的突变(图 2-10)。调控序列突变所产生的遗传学效应,通常可直接体现为蛋白质合成速率的降低或异常增高,进而影响细胞正常的代谢节律,以致引起疾病的发生。而内含子与外显子剪接位点突变,则往往会造成 RNA 编辑错误,以致不能形成正确的 mRNA 分子,这也势必会导致功能蛋白的合成障碍。
移码突变
移码突变(frame-shift mutation)是一种由于基因组DNA多核苷酸链中碱基对的插入或缺失,以致自插入或缺失点之后部分的、或所有的三联体遗传密码子组合发生改变的基因突变形式。移码突变直接的分子遗传学效应就是导致其所编码的蛋白质多肽链中的氨基酸组成种类和顺序的变化。
碱基对插入或缺失的数目、位点不同,对其后密码子组合改变的影响也不尽相同(表2-1)。
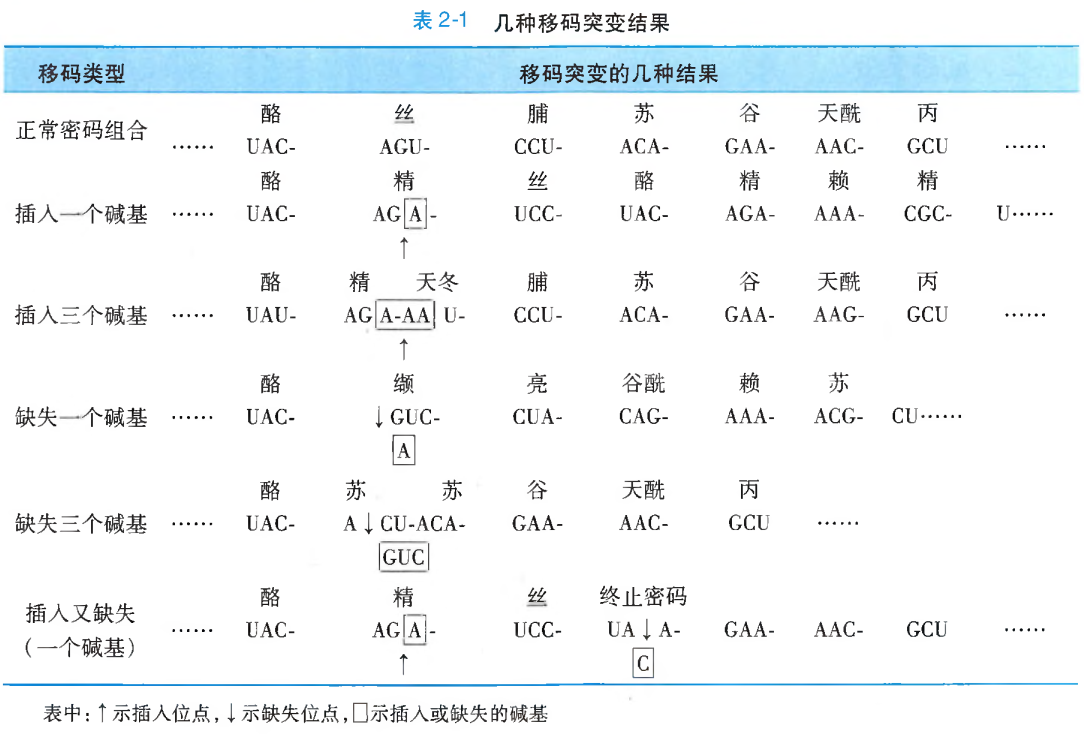
- 一个或两个碱基对的插入或缺失。这将造成插入或缺失位点之后整个密码子碱基组合及其排列顺序的改变;
- 整码突变或框内突变,即如果插入或缺失的碱基对是3或3的倍数,且插入或缺失位点亦恰好在两个相邻的遗传密码子之间,由此所引起的变化是在DNA双链的多核苷酸组成上额外地增加或减少1个或数个三联遗传密码子,但却并不造成
读码框(reading frame)的改变;如果插入或缺失的3碱基对是在同1个三联密码子之内,那就只是造成该插入或缺失位点前、后各1个遗传密码的改变,而并不会改变其他密码子的碱基组成和编码顺序。 - 当在某一位点插入或缺失1~2个碱基对之同时,又在该突变位点之后的某一位点相应地缺失或插入了同样数目的碱基对,那么,除引起前、后两个位点之间的密码组合改变外,其后其他的密码子组合仍可保持正常。
移码突变不仅涉及DNA分子中碱基组成数目的改变,而且还伴随着特定的遗传密码组成性质与排列顺序的改变。因此,所引发的遗传学效应往往是比较严重的。它会导致一条或多条多肽链的合成障碍或功能缺陷,甚至完全丧失,进而危及到机体细胞正常的生命活动。
小片段的缺失、 插入与重排
DNA分子中还可能发生小片段(涉及十几、数十或数百个碱基片段序列)的微小缺失、微小插入或重排。
-
微小缺失。微小缺失(micro-deletion)是由于在DNA复制或损伤的修复过程中,某一小片段没有被正常复制或未能得到修复所致。 -
微小插入。 在DNA的复制过程或损伤过程中,某一小片段插入到DNA链中,其结果造成新链中相应小片段的微小插入(micro-insertion)。 -
重排。重排(rearrangement)发生的分子机制是当DNA分子发生两处以上的断裂后,所形成的断裂小片段两端颠倒重接,或者不同的断裂片段改变原来的结构顺序重新连接,从而形成了重排的片段突变形式。
动态突变
科学家们曾一度认为单基因遗传病主要是由遗传物质在分子水平上发生的点突变所引起,而且,这些突变一般都会在世代传递中保持相对的稳定状态,即上述的静态突变。
直至20世纪80~90年代,随着对人类基因组DNA序列组成及结构特征分析研究的不断深入,才发现某些单基因遗传性状的异常改变或疾病的发生,是由于DNA分子中某些短串联重复序列,尤其是基因编码序列或侧翼序列的三核苷酸串联重复扩增所引起。
因为这种串联三核苷酸的重复次数可随着世代交替的传递而呈现逐代递增的累加突变效应,故而被称之为动态突变(dynamic mutation)。把由动态突变所引起的疾病,统称为三核苷酸重复扩增病(trinucleotide repeat expansion diseases,TREDs)。
遗传多态性
遗传多态性的概念
在同一种群中的某种遗传性状同时存在两种以上不连续的变异型,或同一基因座上两个以上等位基因共存的遗传现象。
作为单一基因座等位基因DNA多样性变异在群体水平的体现,凡是在群体中出现频率大于1%的变异体,无论致病与否,均被称之为遗传多态型;而所有那些出现频率小于1%的变异体,则被称之为稀有变异型(rare variants)。
遗传多态性现象十分普遍。多态性的形成,缘于基因的异变。发生于基因组DNA非编码序列(间隔序列或内含子序列)的变异,一般不会影响基因的结构与功能,也不会产生遗传的表型效应。只有那些位于编码序列和调控序列内的DNA变异,方可产生各种蛋白变异体,或者通过影响RNA的转录,从而导致各种明显的表型差异。对于一个体而言,基因多态性碱基组成序列终生不变,并按孟德尔规律世代相传。
遗传多态性的表现形式
遗传多态性不仅表现为个体水平上的表型遗传性状差异,亦可呈现为细胞水平上染色体遗传的多态性;分子水平上基因组DNA遗传的多态性和蛋白质与酶的多态性以及抗原的多态性等。
个体水平上的表型性状遗传多态性
表型性状遗传多态性是种群中不同个体之间同一遗传性状的表型差异,如人类头发、眼睛的颜色。表型遗传差异的多态性,决定于一组相应的复等位基因的作用。
细胞水平上的染色体遗传多态性
染色体多态性是在种群中经常可见的各种染色体形态的变异。其主要表现为同源染色体大小、形态或染色体带型的改变。此类改变,通常仅涉及染色体的结构异染色质区域,因此,并不表现出显著相关的表型效应。
分子水平上的DNA遗传多态性
人类基因组DNA呈现出多种多样的分子结构和组成形式。依据发现的时序和不同遗传多态性的分子遗传学特征,被分为
限制性片段长度多态性(restriction fragment length polymorphism,RFLP);数目可变的串联重复(variable number tandem repeat,VNTR)多态性;短串联重复序列(short tandem repeat,STR)多态性;单核苷酸多态性(single nucleotide polymorphism,SNP)。
等多种类型。当前,被作为遗传标记而在人类遗传学和医学遗传学相关研究领域中得以广泛应用的主要有两大类。
单核苷酸多态性(SNP)
由基因组DNA序列中单个碱基的转换或颠换所形成的变异;是最简单、最常见、分布最为广泛,也是多态性最为丰富的遗传多态类型之一。研究表明,人类基因组DNA平均约1000bp内就有一个SNP,占已知的人类基因组DNA多态性变异之90%以上。
基因组DNA中任何碱基都有发生变异的可能。因此,SNP既可存在于基因的蛋白编码序列之内,亦可出现在非编码序列之中。存在于蛋白编码序列外显子中的SNP又被称之为编码SNP(coding SNP),简称cSNP。目前,发现的cSNP大约有100000个左右,其变异率仅及非编码序列的20%。就其对遗传性状的表型效应而言,cSNP又被区分为不改变编码蛋白氨基酸序列组成的同义cSNP和可改变氨基酸序列的非同义cSNP两种类别;两者所占比例各为50%。
组成DNA的碱基虽然有4种,但是SNP一般只有2个"等位"成员,呈现为"非此即彼"的"双等位基因"(biallelic)多态性。基于SNP的自身特性,作为一种遗传标记,它常被用来进行对复杂性状与疾病的遗传分析和族群的基因识别以及遗传结构研究。
短串联重复序列多态性(STR)
一类以1~6bp为重复单元,串联重复一到数十次;序列长度小于100bp的DNA结构片段。
STR散在于基因组中各个染色体上,但很少出现在编码DNA序列中。其主要表现为重复序列拷贝数的变异,具有较高的遗传多态性。
基因突变的细胞分子生物学效应
基因突变导致蛋白质功能异常
基因突变对蛋白质结构和功能产生的影响主要表现在以下4个方面:
① 直接影响相关功能蛋白质的生物合成;
② 导致蛋白质产生异常的功能效应;
③ 导致组织细胞中蛋白质表达类型的改变;
④ 涉及蛋白质的分子细胞生物学效应与相应临床表型之间的关系。
通过认识这些分子机制,将有助于从事医学遗传学研究与较为深入地理解基因突变导致遗传病发生的细胞分子生物学效应。
基因突变导致异常蛋白的生成
基因突变是蛋白质发生改变的根本原因,而突变蛋白(mutant protein)的形成则是基因突变的结果和表现形式。基因突变一般通过以下两种机制影响正常蛋白质的合成,导致细胞功能损害并引发疾病:
- 突变影响、干扰了RNA的正常转录以及转录后的修饰、剪辑;或直接改变了由其编码的氨基酸的顺序或构成,从而使其丧失正常功能,即所谓的
原发性损害(primary abnormalities)。 - 突变并不直接影响或改变某一条多肽链的正常氨基酸组成,而是通过干扰该多肽链的翻译过程;或翻译后的修饰、加工;甚至通过对蛋白质各种辅助因子的影响,间接地导致某一蛋白质功能的异常。相对于原发性损害机制,后者被称之为
继发性损害(secondary abnormalities)。
基因突变导致蛋白质功能异常
基因突变导致蛋白质功能异常的表现形式主要有以下几种:即丢失功能、 增强功能、 获得新性状、显性负效应以及异时或异地基因表达等。
丢失功能
丢失功能是最常见的基因突变或基因缺失改变蛋白质功能的表现形式。 基因突变可发生在基因的编码区,也可发生在基因的调节区。 位于编码区的无义突变、移码突变等大多都会导致蛋白质正常功能的丧失,而部分错义突变等可使基因所编码的蛋白质保留部分功能。
获得功能
获得功能是最少见的基因突变改变蛋白质功能的表现形式。 对于一个特定的基因功能而言,并非越强越好。 如果破坏了机体的平衡,获得功能也会造成细胞正常生理功能的紊乱,并最终导致疾病的发生。
获得新性状
有的基因突变会使突变蛋白获得新性状,并赋予突变蛋白致病性。
显性负效应
在一对等位基因中,如果其中一个基因突变,另一个基因正常,即使突变基因的功能完全丧失,理论上仍应保留一半的功能,类似于显性遗传病的杂合子。 但在某种情况下,突变蛋白不仅自身没有生理功能,还会影响另一个正常蛋白质发挥其生理功能,这种由蛋白质相互作用产生的干涉现象称为显性负效应(dominant negative effect)。
显性负效应通常通过蛋白质亚单位形成多聚体的形式实现的。
异时或异地基因表达
有的基因突变影响基因调节区的序列导致该基因在不适当的时间或在不适当的细胞中表达,即所谓异时或异地基因表达。
突变导致组织细胞蛋白表达类型的改变
蛋白质通常可被划分为两类,即持家蛋白(housekeeping protein)和奢侈蛋白(luxury protein)。
持家蛋白存在于几乎所有的组织细胞类型中,为细胞正常结构和最基本的生命活动的维系所必需。如核酸聚合酶蛋白、核糖体蛋白、细胞骨架蛋白等。
奢侈蛋白则仅仅表达、存在于某些特定的组织细胞类型,是特异组织细胞类型分化及特殊生理功能的标志。如B淋巴细胞中的免疫球蛋白。
基因突变往往导致正常组织细胞蛋白表达类型的改变,继而引起细胞功能的异常,甚至发生病理改变。
突变蛋白的分子细胞病理学效应与临床表型之间的关系
同一基因的不同突变产生不同的临床表型
同一基因的不同突变形式往往会导致不同的临床表现,这种现象称为等位基因异质性。
同一基因的不同突变可改变疾病的遗传方式
同一基因在不同位置发生基因突变有时会改变疾病的遗传方式。
基因突变引发“无法预测”的临床效应
遗传病的发生是在一定条件下基因有害突变的必然结果。 然而,在很多情况下却又无法估计和预测到某一基因突变是否能够、或者应该还是不应该引起这样或那样的生理生化异常及与之相应的临床表型效应。
基因突变引起性状改变的分子生物学机制
遗传中心法则阐明了核酸、蛋白质之间的相互关系及细胞内遗传信息的传递、表达过程。
DNA分子中储存的遗传信息,经过转录、翻译传递到肽链。而后者再进一步形成具有生物学功能活性的蛋白质,并最终表现为细胞的结构和功能性状。
这种信息语言的转换和表达过程,不但是基因控制正常遗传性状发育最基本的分子生物学机制,也是基因突变引起各种性状异常和临床疾病发生的基本机制。
基因突变引起酶分子的异常
酶是生物体内具有特殊生物催化活性的催化剂。人体细胞中的每一步生化代谢反应,几乎都需要某种专一性酶的催化才能进行和完成。酶又是基因表达的产物,由结构基因突变所引起的酶分子组成与结构的改变,或由调节基因突变所导致的酶合成异常,都有可能造成相关代谢过程的障碍或代谢程序的紊乱。
如果这种基因突变发生于生殖细胞或受精卵中,就有可能传递给后代,从而产生相应的先天性代谢缺陷(inborn errors of metabolism)或遗传性酶病(hereditary enzymopathy)。
结构基因突变引起的酶蛋白结构异常
酶有单体酶与复合酶之分。前者仅由酶蛋白组成,后者,除酶蛋白外尚含有某种辅基或辅助因子。但无论是何种类型,其催化活性都是建立在与其催化功能相适应的特定三维空间构象基础之上的。
所有结构基因突变,除同义突变一般不会引起酶蛋白结构异常外,其他突变形式都有可能造成酶分子特定立体构象不同程度的改变。空间构象变化引起的酶活性异常,主要表现为以下几种形式:
- 酶的功能活性完全丧失;
- 酶尚具有一定的功能活性,但其稳定性降低,极易被降解而失去活性;
- 酶与其作用底物的亲和性降低,以致不能迅速、有效地与之结合,造成代谢反应的延滞;
- 酶蛋白与辅助因子的亲和性下降,影响了酶的正常活性。
调节基因突变引起的酶蛋白合成异常
基因是一个可调控的遗传功能表达单位。
每一个结构基因的构成,除了其转录序列外,还含有侧翼的非转录调控序列。此类调控序列突变,或者使基因转录的启动发生障碍,不能进行mRNA的合成;或者造成转录速率下降,影响mRNA合成的产量。这些改变最终都导致酶蛋白的缺失,或酶蛋白合成量的不足而引发的代谢缺陷。
酶分子异常引起的代谢缺陷
人体细胞内的绝大多数生理活动都是建立在一系列相互联系的级联生化反应基础之上的。 而在这些级联反应中,每一步几乎都是在特定的酶或酶系的催化下实现和完成的。 因此,酶是实现机体细胞内各种生命活动过程最为直接、极其关键的重要因素之一。
酶与代谢反应的关系
每一个代谢反应途径,以及由此所产生的各种中间代谢产物的最终去向,均和参与催化该代谢反应的酶密切相关。 换言之,即在一定条件下,酶能够决定体内代谢反应的类型和反应的途径及去向。
在体内复杂的代谢反应过程中,参与代谢过程的各种物质,往往表现出作为反应底物和反应产物的双重属性,以及彼此之间互为底物与产物的交错关系。 而这种属性与相互关系,又构成了体内普遍存在的反馈调节机制的基础。
缺陷对代谢反应的影响
酶缺陷造成代谢底物缺乏。绝大多数非脂溶性或极性的小分子物质(如葡萄糖、氨基酸等)都必须依赖于膜转运酶的作用才能进入细胞内作为某种代谢活动的原初反应底物而引发相应的代谢反应。一旦与之相关的膜转运酶缺陷或异常,就会造成代谢底物的缺乏而阻碍和影响整个代谢过程的发生,最终引发一系列的疾病症状。酶缺陷导致代谢产物堆积。酶缺陷导致的代谢产物堆积,可能造成两种情况的发生:堆积产物对机体的直接危害和堆积底物或产物激发代谢旁路开放。酶缺陷导致代谢终产物缺乏。在机体细胞内的物质代谢级联反应中,酶的缺陷出现在其整个过程的任何一个环节或步骤,都可能导致正常反应途径受阻或中断,造成某些必需代谢终产物的缺乏,并引发疾病。酶缺失导致反馈调节失常。在体内一系列级联反应中形成的某些代谢产物,往往会反过来影响、调节其初始的或前一反应步骤的进行以及反应速率,此即所谓的反馈调节。 某些酶的缺陷,若导致此类产物生成的减少或缺失,就可能造成这种自我反馈调节作用的失常,扰乱细胞代谢相对恒定、相互协调的运转秩序,从而引起机体疾病的发生。
非酶蛋白分子缺陷导致的分子病
基因突变除引起酶蛋白分子缺陷而导致代谢性疾病的发生之外,还可以通过影响非酶蛋白分子的结构和数量,从而改变机体细胞的生物学性状,并最终导致机体遗传性状的异常。
因此,一般将由非酶蛋白分子结构和数量的异常所引发的疾病,统称为分子病。比如由某些运输蛋白、免疫蛋白缺陷所引发的疾病,皆属此类。
单基因病的遗传
单基因遗传病(monogenic disease, single-gene disorder),简称单基因病,是由一对等位基因控制而发生的遗传性疾病,这对等位基因称为主基因(major gene)。
单基因遗传病的遗传可分为核基因的遗传和线粒体基因的遗传两种,后者属于细胞质遗传。
核基因遗传的单基因遗传病在上下代之间的传递遵循孟德尔定律,因此也称为孟德尔遗传病,根据致病主基因所在染色体和等位基因间显隐关系的不同,包括五种遗传方式:
- 常染色体显性遗传;
- 常染色体隐性遗传;
- X 连锁显性遗传;
- X 连锁隐性遗传;
- Y 连锁遗传;
染色体显性遗传病的遗传
如果一种遗传病的致病基因位于1 ~22号常染色体上,在杂合子的情况下可导致个体发病,即致病基因决定的是显性性状,这种遗传病就称为常染色体显性(autosomal dominant, AD)遗传病。
常染色体完全显性遗传的特征:
- 由于致病基因位于常染色体上,因而致病基因的遗传与性别无关,即
男女患病的机会均等; - 患者双亲中必有一个为患者,致病基因由患病的亲代传来,此时患者的同胞有 1/2 的发病可能;双亲无病时,子女一般不会患病(除非发生新的基因突变);
- 患者的子代有 1/2 的发病可能;
- 系谱中通常连续几代都可以看到患者,即存在
连续传递的现象;
根据这些特点,临床上可对常染色体显性遗传病进行发病风险的估计。例如,夫妇双方中有一人患病(杂合子),那么子女患病的可能性为 1/2;如果夫妇双方都是患者(均为杂合子),则子女患病的可能性为 3/4。
染色体显性遗传病举例:
| 疾病名称 | 主要症状 | 发病率 |
|---|---|---|
| 软骨发育不全症 | 四肢短小,躯干正常 | 1/15000-1/40000 |
| 多指(趾)症 | 手指或脚趾数目过多 | 1/500-1/1000 |
| 亨廷顿舞蹈症 | 中年发病,运动和认知功能障碍 | 1/10000 |
| 家族性高胆固醇血症 | 血液胆固醇异常升高,易患心血管疾病 | 1/500 |
| 视网膜母细胞瘤 | 儿童眼部恶性肿瘤 | 1/15000-1/20000 |
染色体隐性遗传病的遗传
一种遗传病的致病基因位于常染色体上,其遗传方式是隐性的,只有隐性致病基因的纯合子才会发病,称为常染色体隐性(autosomal recessive, AR)遗传病。
带有隐性致病基因的杂合子本身不发病,但可将隐性致病基因遗传给后代,称为携带者(carrier)。广义地说,携带者是指携带有某种致病基因或异常染色体,但本身并不表现出临床症状的个体,虽然携带者本身并不发病,但可能会将致病基因或异常染色体传递给后代,导致后代发病。
一般认为,常染色体隐性遗传的典型系谱有如下特点:
-
由于致病基因位于常染色体上,因而致病基因的遗传与性别无关,即男女患病的机会均等。
-
患者的双亲表型往往正常,但都是致病基因的携带者;
-
患者的同胞有
1/4的发病风险,患者表型正常的同胞中有2/3的可能为携带者;患者的子女一般不发病,但肯定都是携带者; -
系谱中患者的分布往往是散发的,通常看不到连续传递现象,有时在整个系谱中甚至只有先证者一个患者;
-
近亲婚配(consanguineous marriage)时,后代的发病风险比随机婚配明显增高。这是由于他们有共同的祖先,可能会遗传到同一个隐性致病基因。
常染色体隐性遗传病举例:
| 疾病名称 | 主要症状 | 发病率 |
|---|---|---|
| 白化病 | 黑色素缺乏,皮肤苍白,视力受损 | 1/20000 |
| 镰状细胞贫血症 | 红细胞变形,贫血,易感染 | 1/500(非洲裔) |
| 苯丙酮尿症 | 苯丙氨酸代谢障碍,智力受损 | 1/10000-1/15000 |
| 先天性耳聋 | 听力障碍 | 1/1000-1/2000 |
| 囊性纤维化 | 呼吸和消化系统受损 | 1/2500(白种人) |
| 地中海贫血 | 血红蛋白合成障碍,重度贫血 | 1/50000 |
X连锁显性遗传病的遗传
由性染色体上的基因所决定的性状在群体分布上存在着明显的性别差异。如果决定一种遗传病的致病基因位于X染色体上,带有致病基因的女性杂合子即可发病,称为X连锁显性(X-linked dominant,XD)遗传病。
男性只有一条X染色体,其X染色体上的基因不是成对存在的,在Y染色体上缺少相对应的等位基因,故称为半合子(hemizygote),其X染色体上的基因都可表现出相应的性状或疾病。男性的X染色体及其连锁的基因只能从母亲传来,将来又只能传递给女儿,一般不存在男性→男性的传递,这种传递方式称为交叉遗传(criss-cross inheritance)。
对于X连锁显性遗传病来说,女性有两条X染色体,其中任何一条X染色体上存在致病基因都会发病,而男性只有一条X染色体,所以女性发病率约为男性的2倍。然而男性患者病情较重,而女性患者由于X染色体的随机失活,病情较轻且常有变化。
X连锁显性遗传的典型系谱有如下特点:
- 人群中女性患者数目多于男性患者,在罕见的
XD遗传病中,女性患者的数目约为男性患者的2倍,但女性患者病情通常较轻; - 患者双亲中一方患病;如果双亲无病,则来源于新生突变;
- 由于
交叉遗传,男性患者的女儿全部都为患者,儿子全部正常;女性杂合子患者的子女中有50%的可能性发病; - 系谱中常可看到
连续传递现象,这点与常染色体显性遗传一致;
X染色体显性遗传病举例:
| 疾病名称 | 主要症状 | 遗传特点 | 发病率 |
|---|---|---|---|
| 维生素D抗性佝偻病 | 骨骼发育异常,血磷低,维生素D治疗无效 | 女性患者可生育,子代男女均可能发病 | 1/20000 |
| 色素失禁症 | 皮肤色素沉着,中枢神经系统异常 | 男性胚胎常流产,存活女性表现完整症状 | 1/50000 |
| 视网膜色素变性 | 夜盲,视野缩小,最终可致盲 | 男女均可发病,男性症状较重 | 1/3500 |
| Rett综合征 | 智力发育迟缓,运动功能退化 | 男性胚胎期致死,仅女性患者存活 | 1/10000-1/15000(女性) |
| 低磷性佝偻病 | 骨骼软化,生长发育迟缓 | 男女均可发病,但男性症状更严重 | 1/20000 |
X连锁隐性遗传病的遗传
如果决定一种遗传病的致病基因位于X染色体上,且为隐性基因,即带有致病基因的女性杂合子不发病,称为X连锁隐性(X-Iinked recessive,XR)遗传病。
X连锁隐性遗传的典型系谱有如下特点:
- 人群中男性患者远多于女性患者,在一些罕见的
XR遗传病中,往往只能看到男性患者; - 双亲无病时,儿子有1/2的可能发病,女儿则不会发病,表明致病基因是从母亲传来的;如果母亲不是携带者,则来源于新生突变;
- 由于
交叉遗传,男性患者的兄弟、舅父、姨表兄弟、外甥、外孙等也有可能是患者;患者的外祖父也可能是患者,这种情况下,患者的舅父一般不发病; - 系谱中常看到几代经过女性携带者传递,男性发病的现象;如果存在女性患者,其父亲一定是患者,母亲一定是携带者;
X染色体隐性遗传病举例:
| 疾病名称 | 主要症状 | 遗传特点 | 发病率 |
|---|---|---|---|
| 血友病A | 凝血因子VIII缺乏,易出血,血液凝固障碍 | 男性发病,女性携带 | 1/5000(男性) |
| 血友病B | 凝血因子IX缺乏,症状类似血友病A | 男性发病,女性携带 | 1/30000(男性) |
| 红绿色盲 | 无法分辨红色和绿色 | 男性多见,女性少见 | 8%(男性),0.5%(女性) |
| 杜氏肌营养不良 | 进行性肌肉萎缩,运动功能丧失 | 男孩3-7岁发病,女性携带 | 1/3500(男性) |
| G6PD缺乏症 | 溶血性贫血,对某些药物和食物过敏 | 男性症状明显,女性多为携带者 | 1/1000(男性) |
Y连锁遗传病的遗传
如果决定某种性状或疾病的基因位于Y染色体,随Y染色体而在上下代之间进行传递,称为Y连锁遗传(Y-Iinked inheritance)。
Y连锁遗传的传递规律比较简单,具有Y连锁基因者均为男性,这些基因将随Y染色体进行父→子→孙的传递,因此又称为全男性遗传(holandric inheritance) 。
目前已经定位在Y染色体上的基因有54个,其中主要的有睾丸决定因子基因(SRY,OMlM *480000)和外耳道多毛症基因(OMlM 425500)等。
从性遗传和限性遗传
从性遗传(sex-influenced inheritance)是位于常染色体上的基因,由于受到性别的影响而显示出男女表型分布比例的差异或基因表达程度的差异。
例如,雄激素性秃发1型属于常染色体显性遗传,群体中男性患者明显多于女性。男性杂合子(Aa)即会出现秃顶,表现为从头顶中心向周围扩展的进行性、弥漫性、对称性脱发,仅枕部及两侧颞部保留头发;而女性杂合子(Aa)仅表现为头发稀疏而不会表现秃顶症状。出现这种情况是因为雄激素性秃发(AGIA)基因的表达会受到体内雄性激素的影响。但携带有AGIA基因的女性杂合子,由于某种原因导致体内雄性激素水平升高也可出现秃顶的症状。
限性遗传(sex-limited inheritance)则指位于常染色体上的基因,由于基因表达的性别限制,只在一种性别表现,而在另一种性别则完全不能表现,但这些基因均可传给下一代。限性遗传可能主要是由于男女性在解剖学结构上的差异所致,也可能受性激素分泌方面的性别差异限制,故只在某一性别中发病,如女性的子宫阴道闭水,男性的尿道下裂等。
多基因病的遗传
与单基因遗传疾病的罕见性不同,多基因疾病多为常见病且表型取决于相关的多个基因的共同作用。这些基因对疾病的表型贡献有大有小,因此可分为主效基因(major effect gene)和微效基因(minor effect gene)。
主效基因可能存在显、隐性关系,但微效基因相互之间显隐之分并不明确,多互为共显性。多对微效基因的作用积累之后,可以形成一个明显的效应,这种现象称为累加效应(additive effect);因而这些基因也被称作累加基因(additive gene)。
近年来的研究发现,微效基因所发挥的作用或者说贡献率并不是等同的,可能存在一些起主要作用的所谓主基因(major gene),主基因有可能存在显、隐性关系。由于多基因疾病参与的基因多,不仅基因之间遗传关系复杂,同时,这类疾病还往往明显受环境影响,因此,这类性状也称为复杂性状,这类疾病也就称为复杂疾病。
数量性状的多基因遗传
数量性状与质量性状
在单基因遗传中,基因和表型之间的对应关系较为明显,因此基因改变而引起的性状的变异在群体中的分布往往是不连续的,可以明显地分为若干群,基本与其基因型相对应,且性状不易受环境影响,所以单基因遗传的性状也称为质量性状。
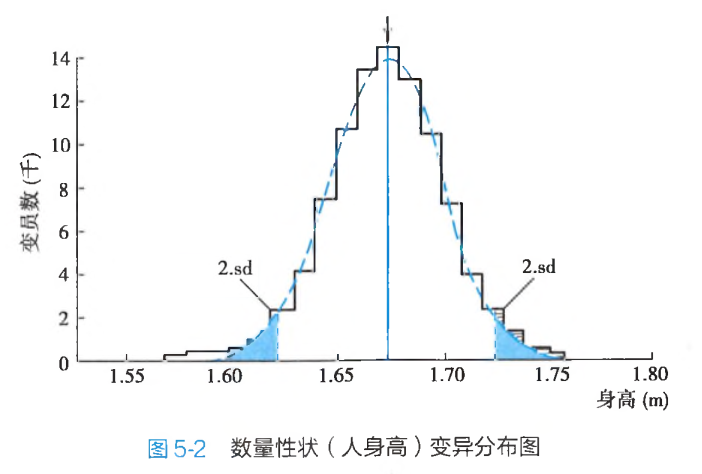
多基因遗传性状的变异在群体中的分布是连续的,只有一个峰,因此会有一个平均值。不同个体间的差异只是量的变异,临近的两个个体之间的差异很小,因此这类形状称为数量性状(quantitative character)。
因此,数量性状是一种可测量的生理或生化数值指标,如身高、体重、血压、血清胆固醇浓度或体重指数等。群体中,每个个体在这些数量性状的数值上存在差别,呈现由低到高逐渐过渡。数值极高或极低的个体只占少数,大部分个体数量性状数值接近平均值。将此数量性状变异分布绘成曲线,该曲线往往表现出正态分布(图5-2)。这些性状在人群中呈常正态分布,而非"有或无"的遗传方式。
数量性状的多基因遗传
多基因遗传中,虽然性状的遗传规律不符合孟德尔定律,但每一对基因的遗传方式仍符合孟德尔定律,即分离和自由组合。
对于某一个数量性状而言,每个个体的控制基因数量是基本相同的,但各型基因的比例是不同的,因而造成性状具有差异性。一般说来,决定数量性状的基因远不止3对,而且许多研究也显示每个基因的作用也并非相等。
疾病的多基因遗传
易患性与发病阈值
在多基因遗传病中,遗传基础是由多基因构成的,它部分决定了个体发病的风险。这种由遗传基础决定一个个体患病的风险称为易感性(susceptibility)。由于环境对多基因遗传病产生较大影响,因此学术界将遗传因素和环境因素共同作用决定个体患某种遗传病的风险称为易患性(liability),也就是说易感性+环境因素=易患性。
在相同环境下不同个体产生的差异,可以认为是由不同的易感性造成的,也就是说是基因差异造成的。一般群体中,易患性很高或很低的个体都很少,大部分个体都接近平均值。因此,群体中的易患性变异也呈正态分布。但在一定的环境条件下,易感性高低可代表易患性高低。当一个个体易患性高到一定限度时,就可能发病。这种由易患性所导致的多基因遗传病发病最低限度称为发病阈值(threshold)。阈值将连续分布的易患性变异分为两部分:正常群体和患病群体(图5-4)。因此,多基因遗传病又属于阈值相关疾病,阈值是易患性变异的某一点,在一定条件下,阈值代表患病所必需的、最低的易患基因的数量。
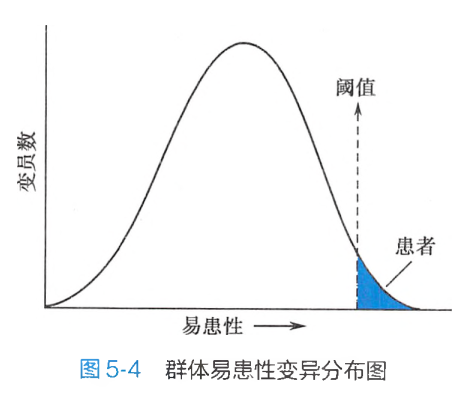
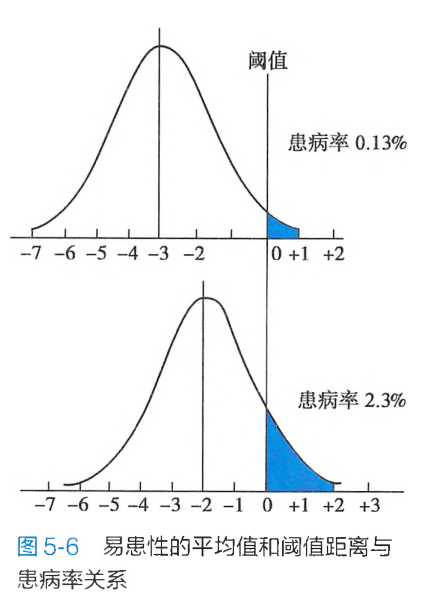
一种多基因病的易患性的平均值与阈值越近,表明易患性高,阈值低,群体患病率高。相反,易患性的平均值与阈值越远,表明易患性低,阈值高,群体患病率低(图5-6)。
遗传率及其估算
多基因遗传病是遗传因素和环境因素共同作用所致。
这其中,遗传因素的作用大小可用遗传率来衡量。遗传率(heritability)又称遗传度,是在多基因疾病形成过程中,遗传因素的贡献大小。遗传率愈大,表明遗传因素的贡献愈大。
如果一种疾病完全由遗传因素所决定,遗传率就是100%;如果完全由环境所决定,遗传率就是0,这两种极端情况是极少见的。某些疾病的遗传率较高,可达70%~80%,这表明在决定疾病易患性变异上,遗传因素发挥了较大的作用,相对环境因素的作用较小;某些疾病的遗传率较小,仅为30%~40%,这表明在决定疾病易患性变异上,环境因素发挥了较大作用,相对遗传因素的作用较小。
一般说来,遗传率越低的性状或疾病,家族聚集现象越不明显。
计算人类多基因遗传病遗传率的高低在临床实践上有重要意义,传统的计算方法主要有两种,即Falconer公式和Holzinger公式。
Falconer公式
Falconer公式(Falconer method)是根据先证者亲属的患病率与遗传率有关而建立的。亲属患病率越高,遗传率越大,所以可通过调查先证者亲属患病率和一般人群的患病率,算出遗传率(或H)。
(式5-1)
式5-1中,为遗传率;为亲属易患性对先证者易患性的回归系数;为亲缘系数。
当已知一般人群的患病率时,用式5-2计算回归系数:
(式5-2)
当缺乏一般人群的患病率时,可设立对照组,调查对照组亲属的患病率,用式5-3计算回归系数:
(式5-3)
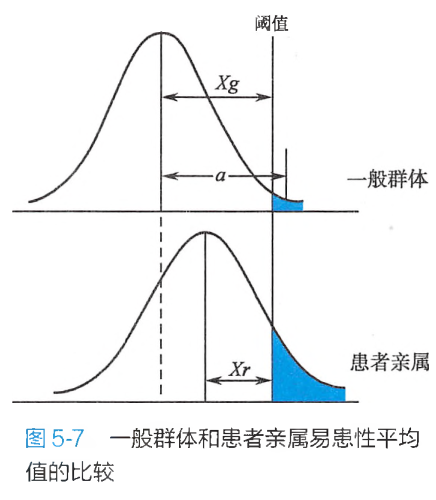
在式5-2和式5-3中,为一般群体易患性平均值与阈值之间的标准差数;为对照组亲属中的易患性平均值与阈值之间的标准差数;为先证者亲属易患性平均值与阈值之间的标准差数;为一般群体易患性平均值与一般群体中患者易患性平均值之间的标准差数(图5-7);为先证者亲属易患性平均值与先证者亲属中患者易患性平均值之间的标准差数;为一般群体患病率;为对照亲属患病率,;为先证者亲属患病率。
、、和均可由一般群体患病率、对照亲属患病率和先证者亲属患病率查Falconer表得到。
亲缘系数是指两个个体从共同祖先获得某一特定等位基因的总体概率,可见Falconer表。
在亲缘系数中,一级亲属指一个人与其双亲、子女和同胞之间,其基因有1/2的可能性是相同的;二级亲属指一个人与其叔、伯、姑、舅、姨、祖父母和外祖父母之间,其基因有1/4的可能性是相同的;三级亲属指一个人与其表兄妹、堂兄妹、曾祖父母之间,其基因有1/8的可能性是相同的。
Falconer公式应用
先天性房间隔缺损在一般群体中的患病率为1/1000(0.1%),在100个先证者的家系中调查,先证者的一级亲属共有669人(双亲200人,同胞279人,子女190人),其中有22人发病。
依次求得先证者一级亲属的患病率为22/669×100%=3.3%(),然后查Falconer表。按群体患病率查得和,再根据亲属患病率查得和,然后代入式5-2求出值。
将值代入式5-1:
以上计算结果表明,遗传因素对先天性房间隔缺损发生的贡献为74%,经显著性检验该遗传率有统计学意义。
在缺乏一般人群患病率数据时,可选择与病例组匹配的对照组,调查对照组亲属的患病率,用先证者亲属和对照亲属的患病率计算遗传率。
对江苏启东肝癌的调查发现,肝癌患者一级亲属6591人中,有359人发病,其患病率为5.45%();在年龄和性别均与患者相应的无病对照者的5227名一级亲属中,有54人患肝癌,患病率。,分别查得、和、,然后代入式5-3求出值。
将值代入式5-1:
以上计算结果表明,遗传因素对肝癌发生的贡献超过50%,经显著性检验该遗传率有统计学意义。
Holzinger公式
Holzinger公式(Holzinger formula)(1929)是根据遗传率越高的疾病,单卵双生的患病一致率与二卵双生患病一致率相差越大而建立的。
单卵双生(monozygotic twin,MZ)是由一个受精卵形成的一对双生子,他们的遗传基础理论上是完全相同的,其个体差异主要由环境决定;二卵双生(dizygotic twin,DZ)是由两个受精卵形成的一对双生子,相当于同胞,因此他们的个体差异由遗传基础和环境因素共同决定。
所谓患病一致率是指双生子中一个患某种疾病,另一个也患同样疾病的频率。其中,为单卵双生子的同病率;为二卵双生子的同病率。
(式5-4)
Holzinger公式应用
举个例子,对躁狂抑郁性精神病的调查表明,在15对单卵双生子中,共同患病的有10对;在40对二卵双生子中,共同患病的有2对。依此来计算单卵双生子的同病率为67%,二卵双生子的同病率为5%。代入式5-4:
以上结果表明,在躁狂抑郁性精神病中,遗传因素的贡献为65%。
常见多基因遗传病的患病率和遗传率
群体遗传
群体(population)又称种群,是属于一个物种,生活在同一地区,并且能够相互杂交的个体群。群体是物种的基本结构单位。群体中进行有性生殖的所有个体所拥有的基因型构成基因库(gene pool)。基因库即一个群体中所包含的全部遗传信息,含有特定位点的全部等位基因。
群体遗传学(population genetics)研究群体的遗传变异分布,特别是等位基因频率和基因型频率在人群中的维持、变化及其规律。
遗传病在不同种族或民族人群中的差异和变化规律,也属于群体遗传学研究的重要范畴。群体遗传学应用于医学,是要探讨遗传病或复杂性状在人群中的遗传方式、致病基因频率及其变化的规律,开发相应的遗传统计方法,故又称之为遗传流行病学(genetic epidemiology)。
群体的遗传平衡
Hardy-Weinberg 平衡定律
按照孟德尔遗传规律,某一性状由一对等位基因决定,可分别标识为 A 和 a,等位基因在人群中的分布频率,称为等位基因频率(allele frequency)。而这一对等位基因组成 3 种可能的基因型(genotype),分别为 AA、Aa 和 aa。对于人群中的任一个体,其基因型只能为 AA、Aa 和 aa 之一。基因型在人群中分布的频率,称为基因型频率(genotype frequency)。
对于常染色体显性遗传病,纯合子 AA 和杂合子 Aa 显现相同的表型;而对于隐性遗传病,纯合子 AA 与杂合子 Aa 及纯合子 aa 呈现不同的表型。由于基因型无法直接进行观察,过去多用表型频率来推测基因型频率。但这需要满足 2 个条件:①单基因遗传;②不同的基因型与表型一一对应。有些性状虽然符合单基因遗传方式,但表型与基因型并不一一对应。例如,ABO 血型的每种表型对应几十甚至上百种基因型。随着 DNA 测序和基因分型方法的快速发展,基因型的获取已不存在困难。
例如,在一个 747 人的人群中,某个 SNP 位点 AA 基因型的频率(假设为 D)是 31.2%,AG 基因型的频率(假设为 H)是 51.5%,GG 基因型(假设为 R)为 17.3%。则等位基因 A 的频率(设为 p)为
等位基因 G 的频率(设为 q)为
即等位基因频率 p、q 与基因型频率 D、H、R 的关系为:
对于一个群体,一个单基因遗传的性状,其表型由基因型频率决定。
那么,这个群体的表型频率会怎样变化?等位基因频率和基因型频率的关系又是什么?
群体遗传学的核心概念是Hardy-Weinberg 平衡定律(Hardy-Weinberg law)。该定律解释了等位基因频率与基因型频率的关系,并在一定条件下,群体的等位基因频率和基因型频率在向子代传递的过程中保持不变。
一对等位基因 A 和 a,其等位基因频率分别为 p 和 q,,则群体的基因型频率为 的二项式展开:。其中,、 和 分别为基因型 AA、Aa 和 aa 的频率。这就是 Hardy-Weinberg 定律的内涵。
Hardy-Weinberg 平衡的成立必须满足以下几个条件:
- 群体无限大;
- 群体内的个体随机交配;
- 没有自然或人工选择;
- 没有突变;
- 群体内没有大规模的个体迁移;
可以说,没有完全满足 Hardy-Weinberg 平衡成立条件的群体,但一个足够大的群体在一定时间内应该近似地被看作一个遗传平衡群体。
对于复等位基因,即多于 2 个等位基因的位点,Hardy-Weinberg 平衡依然成立。任何纯合子的频率等于等位基因频率的平方,而杂合子频率等于 2×等位基因频率之积。如 3 个等位基因(p、q、r)的位点:
由于男性只有一个 X 染色体,X 连锁是一个特例。男性的基因型频率=等位基因频率,而女性的基因型和等位基因频率与常染色体等位基因相同。
Hardy-Weinberg 定律的应用
遗传平衡群体的判定
针对一个群体的某一特定位点,可以从基因型频率来判断该群体是否在该位点达到遗传平衡。
首先,可以通过基因型频率()的观察值(O)计算出等位基因频率(p、q);再由等位基因频率(p、q)按照 计算出基因型频率的期望值(E);再进行卡方检验:
(式 6-1)
其中,O 和 E 分别为基因型频率的观察值和期望值。
例如,在一个 730 个体的人群中,对一个 A/G 进行基因分型,得到 AA、AG 和 GG 基因型的人数分别为 22、216 和 492 例。因而观察到的基因型频率分别为 0.03、0.296 和 0.674。由此可计算出等位基因频率:
则基因型的期望频率:
AA、AG、GG 的期望值(E):
代入式 6-1,则
以自由度 n=1,得出 P=0.00049,此群体的等位基因频率和基因型频率分布不符合Hardy-Weinberg 平衡。
一般来说,在一个正常的大群体中,人类基因组的任何位点都应该达到 Hardy-Weinberg 平衡,特别是此位点的等位基因 A 的频率高达 17.8%,因而该位点的基因分型很可能存在错误。
虽然从理论上说,人类基因组任何位点都应该符合 Hardy-Weinberg 平衡,但有些位点存在强烈的自然选择(如镰状细胞贫血症),或小群体对大群体的等位基因频率有影响(如ABO血型),使得某些特别位点存在不符合 Hardy-Weinberg 平衡的现象。
基因频率的计算
对于单基因病,当已知一个性状在某群体中的频率,根据 Hardy-Weinberg 平衡的等位基因频率和基因型频率的关系,即可确定等位基因频率和杂合子频率。
例如,某常染色体隐性遗传病在某群体的发病率为 1/10000,该群体的致病基因携带者的频率是多少?
由 ,得出 ,。
故致病基因携带者的频率为:
上述疾病患儿的双亲为肯定携带者。若他们离异后与群体中的任一个体再婚,假设新配偶的家族中无相同疾病的家族史,再生出患儿的风险=(肯定携带者的风险)×(新配偶为携带者的风险)×1/4=。
例如,
囊性纤维化(OMIM#219700)是一种常染色体隐性遗传病,在欧洲白色人种中的发病率约为 1/2000,预测白色人种中囊性纤维化突变基因携带者是多少?
,则 ,。
致病基因携带者的频率为:。
由于白色人种中约有 4% 为囊性纤维化致病基因携带者,这些携带者的生存和婚配是囊性纤维化致病基因传递下去的重要原因,该数据对囊性纤维化家族的遗传咨询十分重要。
对于罕见的隐性遗传病(),p 近似于 1,故杂合子频率()约为 ,即杂合子频率是致病等位基因频率(q)的 2 倍;因此,群体中致病基因携带者的人数()远高于患者()。随着隐性遗传病发病率()的下降,携带者和患者的比率明显升高,这对于制定隐性遗传病的筛查计划具有重要意义。
X 连锁基因频率的估计不同于常染色体基因。因为男性为半合子,男性发病率等于致病等位基因频率 q。
对于一种相对罕见的 X 连锁隐性遗传病(如血友病 A),其男性发病率为 1/5000,则该群体致病等位基因频率 ;女性携带者频率 ,女性发病率为 ,因而男性患者远高于女性患者的发病率。
相反,对于 X 连锁显性遗传病,男性发病率是女性发病率()的 1/2。
例如,X 连锁隐性遗传病
红绿色盲在英国有 1/12 的男性受累。女性是携带者的比例是多少?受累女性的比例是多少?
已知:。
女性致病等位基因携带者的频率:。
女性患者:。
影响遗传平衡的因素
前已述及,Hardy-Weinberg平衡适用的条件包括群体无限大、随机婚配、无突变、无选择、无迁移等。 但是,真正的随机婚配和无限大群体并不存在。 群体越小,群体的等位基因频率受非随机婚配、选择、 迁移等的影响越明显。 下面就讨论群体遗传平衡的因素。
非随机婚配
在4代之内有共同的祖先者均属近亲,如果他们之间进行婚配就成为近亲婚配。亲属关系的远近可用亲缘系数表示,它是指有亲缘关系的两个个体携带相同基因的概率。
如父母与子女和同胞兄弟姐妹之间都各有1/2的基因相同,他们之间的亲缘系数为1/2,父母、兄弟姊妹也被称为一级亲属;与祖父母、 外祖父母、 叔、 姑、 舅、 姨、 侄、 甥的亲缘系数是1/4 ,为二级亲属;表兄妹、堂兄妹之间的亲缘系数是1/8,为三级亲属。
如果发生近亲结婚,夫妇双方均有可能从共同祖先遗传到同一等位基因,并把该等位基因传递给他们的子女,使子女成为该基因的纯合子。
突变和选择
突变是群体发生变异的根源。基因突变对于群体遗传组成的改变有 2 个重要的作用:
- 突变本身改变了基因频率;
- 突变又为选择提供了材料。
突变和选择的交互作用,构成了生物进化的遗传学基础。
选择主要是通过增加和减少个体的适合度来影响基因平衡。
换言之,当一个群体的不同个体的适合度(fitness,f)不同时,选择就会发生作用。自然选择(natural selection)和人工选择(artificial selection)都是导致基因频率变化的重要因素。就人类而言,导致基因频率变化的主要选择因素是自然选择。
遗传漂变
小群体或隔离人群中基因频率的随机波动称为遗传漂变(genetic drift)。由于群体较小,故等位基因在传递过程中会使有的基因固定下来而传给子代,有的基因则丢失,最终使得该基因在群体中消失。遗传漂变的速率取决于群体的大小。群体越小,漂变的速率越快,常常在几代甚至一代后即可出现基因的固定和丢失。
在一个大群体中,如果没有突变发生,则根据Hardy-Weinberg平衡定律,不同基因型的频率将会维持平衡状态。但在一个小群体中,由于与其他群体相隔离,不能够充分地随机交配,故小群体内的基因不能达到完全分离和自由组合,造成基因频率容易产生偏差,但这种偏差不是由于突变、选择等因素引起的。
迁移和基因流
迁移(migration)又称移居,即不同人群的流动和通婚,彼此渗入外来基因,导致基因流动,可改变原来群体的基因频率,这种影响称为迁移压力。
迁移压力的增强可使某些基因从一个群体有效地散布到另一个群体,称为基因流(gene flow)。
注意点
影响遗传平衡的因素并不是独立存在的,群体越小,突变、选择、遗传漂变、非随机婚配的影响就越明显。 影响遗传平衡的因素对规模较大的群体基因频率的影响较小,可以观察到的表型变化相对不明显。
连锁不平衡及其应用
在人类基因组中存在着大量的序列变异,其中在群体中能够以孟德尔遗传的方式传递到子代的变异称为DNA多态。这些变异以单核苷酸多态性(SNP)最为常见。这些多态性多数并不影响个体的表型。
连锁不平衡(linkage disequilibrium)是指不同位点上各等位基因在群体中的非随机组合,即不同基因座上的各等位基因一起遗传到子代的频率明显高于其随机传递的频率。如图6-7所示,某致病突变发生之后,由于发生重组,离该致病位点越近的区域,越容易被一起传递到子代。经过多代之后,与致病基因位点一起传递下来的区域变得很小。由于该位点及其周围区域来源于若干代前的同一段染色体区域,这段区域的各个多态性位点之间即存在连锁不平衡。

例如,两个相邻的SNP位点,分别为A/G和G/T多态。由于这两个位点之间存在连锁不平衡,单倍型(haplotype)A-G总在一起被传递到子代。如果一旦在这两个位点之间发生重组,A-G的单体型就被破坏。
两个位点之间的连锁不平衡程度常用D’或r²来度量,当D’和r²=1时为完全连锁不平衡,一般认为两个位点间r²>0.8时存在明确的连锁不平衡。
一般来说,存在连锁不平衡的区域总是比较小的,在100kb以内。在特定人群中,某一段存在连锁不平衡的区域源于同一祖先。
全基因组关联分析(genome wide association study,GWAS)
GWAS,利用高通量的基因分型手段获得覆盖基因组的SNP基因型,从而进行基因型-表型的关联分析。
对GWAS结果的解释需要注意以下几点:
- 对疾病或性状存在显著关联的SNP位点并不代表功能上的联系,只是说明该SNP与致病基因位点间可能存在连锁不平衡(除非SNP本身就是致病突变,或与致病基因的表达有关);
- 要注意
多重检验的调整,100万个SNP关联分析的显著性差异水平应该是P<5×10⁻⁸(0.05/1×10⁶); - 对于复杂性状,每个易感基因位点的遗传相对风险(genetic relative risk,GRR)可能并不高,需要较大的样本量才能保证检验效能(power)。
线粒体病的遗传
人类线粒体基因组
mtDNA是核基因组外的一独立的基因组。线粒体基因组的结构:
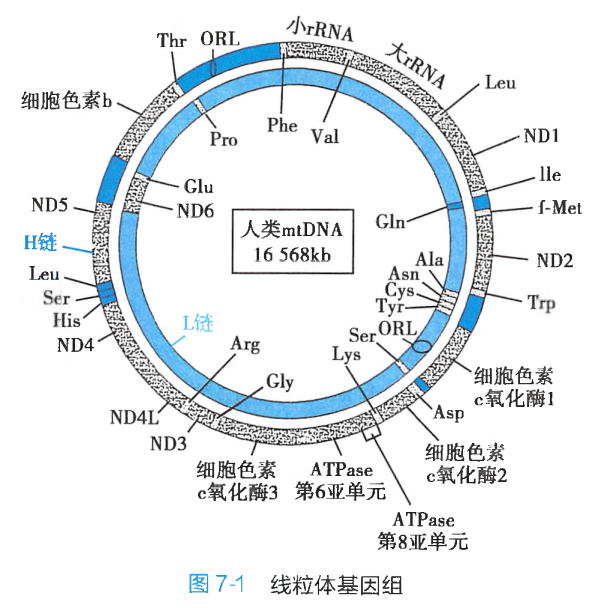
mtDNA可进行半保留复制。与核基因转录比较,mtDNA的转录有以下特点:
- 两条链均有编码功能;
- 两条链从D-环区的启动子处同时开始以相同速率转录,L链按顺时针方向转录,H链按逆时针方向转录;
mtDNA的基因之间无终止子,因此两条链各自产生一个巨大的多顺反子初级转录产物;mtDNA的遗传密码与核基因组(nDNA)不完全相同:UGA编码色氨酸而非终止信号,AGA、AGG是终止信号而非精氨酸,AUA编码甲硫氨酸兼启动信号,而不是异亮氨酸的密码子;
线粒体遗传系统的特点
半自主性
mtDNA能够独立地复制、 转录和翻译,但这种自主性有限。
同质性和异质性
线粒体mtDNA的多质性是区别于核DNA的重要特性。
一个人体细胞内通常有数百个线粒体,每个线粒体内含2~10个mtDNA分子(例外的是血小板和未受精的卵子,它们中的每个线粒体内只含有一个拷贝的mtDNA),所以每个细胞有数千个mtDNA分子,这即为mtDNA的多质性(polyplasmy)。
多质性是线粒体DNA遗传异质性和同质性的基础。
- 细胞或组织中,如果所有
mtDNA分子都是相同的,则称为同质性(homoplasmy)。 - 由于
mtDNA随机突变会产生部分突变型的mtDNA,导致同一个体不同组织、同一组织不同细胞、同一细胞的不同线粒体,甚至同一线粒体内有不同的mtDNA拷贝,这称为异质性(heteroplasmy)。
不同的遗传密码
在mtDNA遗传密码中,有4个密码子的含义与通用密码(nDNA的遗传密码)不同。
母系遗传
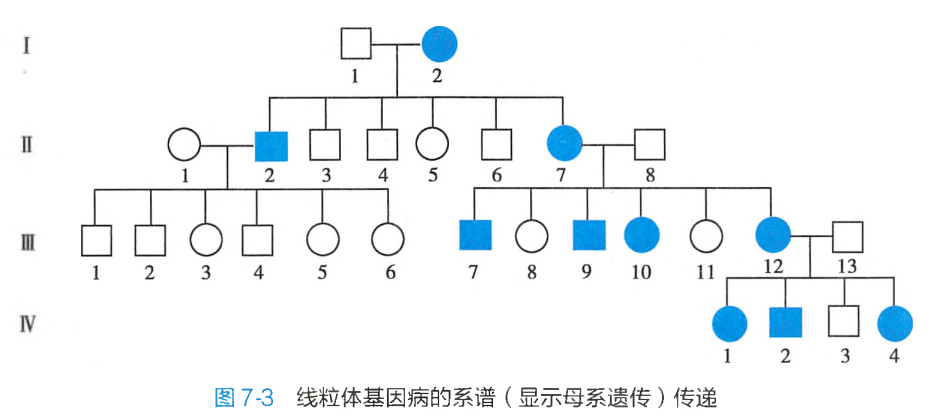
在精卵结合时,卵母细胞拥有上万万拷贝的mtDNA,而精子中只有很少的线粒体,受精时精子中的线粒体几乎不进入受精卵,因此,受精卵中的线粒体DNA几乎全都来自于卵子,这种受精过程中细胞质行为决定了线粒体遗传病的传递方式不符合孟德尔遗传,而是表现为母系遗传(maternal inheritance),即母亲将mtDNA传递给她的子女,但只有女儿能将其mtDNA传递给下一代(图7-3)。
复制分离
细胞分裂时,突变型和野生型mtDNA发生分离,随机地分配到子细胞中,使子细胞拥有不同比例的突变型mtDNA分子,称为复制分离。这种随机分配导致子细胞中mtDNA种类和比例变化,在连续的分裂过程中,子代细胞中突变型mtDNA和野生型mtDNA的比例会发生漂变,向纯质的方向发展。
mtDNA突变率高
mtDNA的结构特点决定了其突变率高的特点,mtDNA突变率比nDNA高10 ~20倍。
线粒体基因突变及相关疾病
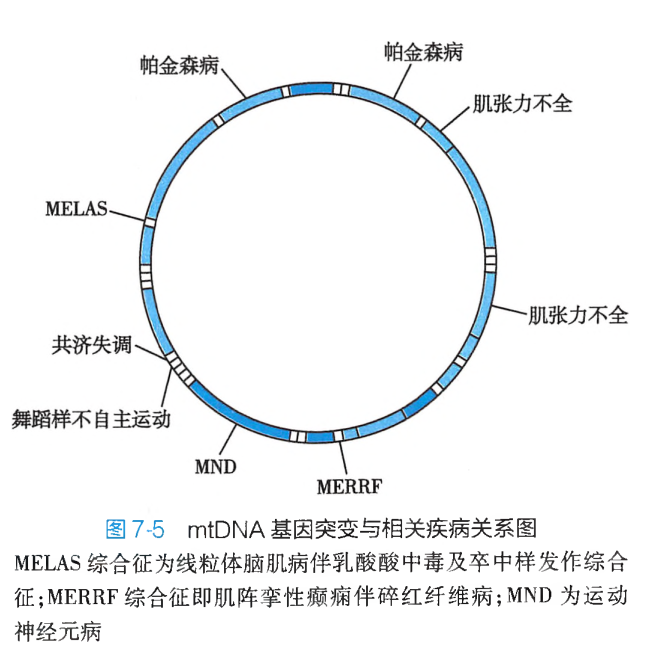
目前,已发现100多个与疾病相关的mtDNA点突变、200多种mtDNA缺失和重排。
由于mtDNA基因突变可影响线粒体氧化磷酸化功能,使ATP合成减少,所以mtDNA突变导致的线粒体疾病多累及能量需求旺盛的肌肉和中枢神经组织,一旦线粒体不能提供足够的能量则可引起细胞退变甚至坏死,导致这些组织和器官功能的减退,出现相应的临床症状(图7-5)。
线粒体疾病的遗传特点
母系遗传
线粒体基因组存在于细胞质中,精卵结合形成的受精卵中细胞质来自卵母细胞,因此只有母亲的线粒体疾病可遗传给子女,而父亲的线粒体疾病不会遗传给后代,称为母系遗传(见图7-3)。但由于受精卵成熟过程中只有一小部分线粒体成熟并通过细胞分裂传给子细胞,加之细胞分裂过程中的复制分离和遗传漂变现象,所以并非女性患者的后代全部发病,而且发病年龄也不一致;甚至一些女性患者本身表型正常,但可将本病传给下一代。
线粒体疾病母系遗传与常染色体病的X连锁遗传不同,前者只能由母亲传递给儿子和女儿,且只有女儿能传递给下一代,男性患者是不会传递给下一代的;而后者中男女患者都可以将疾病传递给下一代,且存在交叉遗传。
阈值效应
mtDNA突变表型由野生型与突变型mtDNA的相对比例以及该组织对能量的依赖程度决定的。
通常突变的mtDNA达到一定数量时,才引起某种组织或器官的功能异常,这种能引起特定组织器官功能障碍的突变mtDNA的最小数量称为阈值。
核质协同性
线粒体疾病受线粒体基因组和核基因组两套遗传系统共同控制,表现为核质协同作用的特点。
人类染色体
人类染色体的基本特征
染色质和染色体
染色质(chromatin)和染色体实质上是同一物质在不同细胞周期、执行不同生理功能时不同的存在形式。
在细胞从间期到分裂期过程中,染色质通过螺旋化凝缩(condensation)成为染色体,而在细胞从分裂期到间期过程中,染色体又解螺旋舒展成为染色质。
染色质
染色质是间期细胞核中伸展开的 DNA 蛋白质纤维。间期细胞核的染色质可根据其所含核蛋白分子螺旋化程度以及功能状态的不同,分为常染色质(euchromatin)和异染色质(heterochromatin)。
常染色质和异染色质。常染色质在细胞间期螺旋化程度低,呈松散状,染色较浅而均匀,含有单一或重复序列的 DNA,具有转录活性,常位于间期细胞核的中央部位。异染色质在细胞间期螺旋化程度较高,呈凝集状态,而且染色较深,多分布在核膜内表面,其 DNA 复制较晚,含有重复 DNA 序列,很少进行转录或无转录活性,是间期核中不活跃的染色质。性染色质。 性染色质(sex chromatin)是性染色体(X 和 Y)在间期细胞核中显示出来的一种特殊结构,包括 X 染色质和 Y 染色质。
染色体
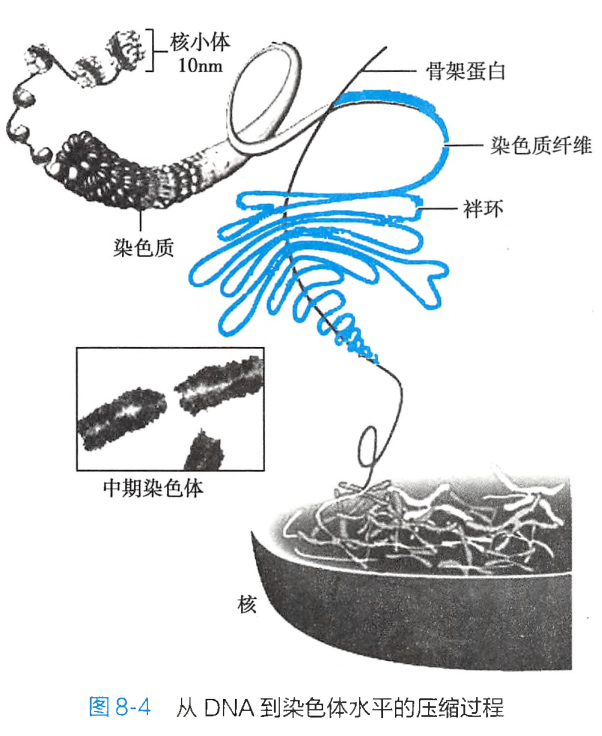
染色质由无数个重复的核小体(nucleosome)单位构成。
核小体则由 4 种组蛋白(H2A、H2B、H3、H4各2个分子)组成的八聚体核心表面围以长约 146bp 的 DNA双螺旋 所构成:
人类染色体的数目、 结构和形态
不同物种生物的染色体数目、结构和形态各不相同,而同一物种的染色体数目、结构和形态则是相对恒定的。
人类染色体的数目
在真核生物中,一个正常生殖细胞(配子)中所含的全套染色体称为一个染色体组,其所包含的全部基因称为一个基因组(genome)。
具有一个染色体组的细胞称为单倍体(haploid),以 n 表示;具有两个染色体组的细胞称为二倍体(diploid),以 2n 表示。人类正常体细胞染色体数目是 46,即 2n=46 条,正常生殖细胞(精子或卵子)中染色体数为 23 条,即 n=23 条。
人类染色体的结构、形态
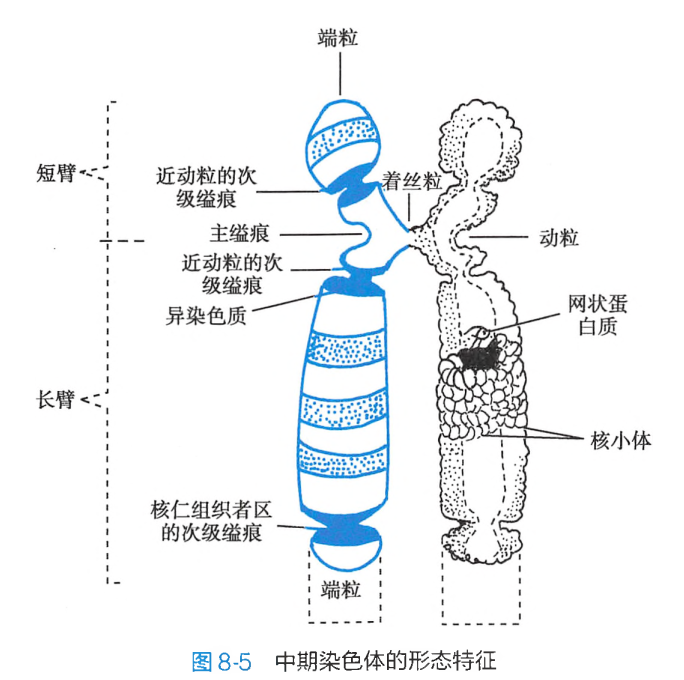
每一中期染色体都具有两条染色单体(chromatid),互称为姊妹染色单体,它们各含有一条 DNA 双螺旋链。两条单体之间由着丝粒(centromere)相连接,着丝粒处凹陷缢窄为初级缢痕或主缢痕(primary constriction)。
着丝粒是纺锤体附着的部位,在细胞分裂中与染色体的运动密切相关,失去着丝粒的染色体片段通常不能在分裂后期向两极移动而丢失。着丝粒将染色体划分为短臂(p)和长臂(q)两部分。在短臂和长臂的末端分别有一特化部位,称为端粒(telomere)。端粒起着维持染色体形态结构的稳定性和完整性的作用。
性别决定及性染色体
人类性别是由细胞中的性染色体所决定的。
在人类的体细胞中有23对染色体,其中22对染色体与性别无直接关系,称为常染色体(autosome)。
常染色体中的每对同源染色体的形态、结构和大小都基本相同;而另外一对与性别决定有明显而直接关系的染色体称为性染色体(sex chromosome),其中包括X染色体和Y染色体。
两条性染色体的形态、结构和大小都有明显的差别。男性的性染色体组成为XY,而女性的性染色体组成为XX,即男性为异型性染色体,女性为同型性染色体。这种性别决定方式为XY型性别决定。人类的性别是精子和卵子在受精的瞬间决定的,确切地说是由精子决定的。在自然状态下,不同的精子与卵子的结合是随机的,因而人类的男女比例大致保持1:1。
很显然,性别是由精子中带有的X染色体或Y染色体所决定的,而X染色体和Y染色体在人类性别决定中的作用并不相等。一个个体无论其有几条X染色体,只要有Y染色体就决定男性表型(睾丸女性化患者除外)。
染色体畸变
染色体畸变可分为数目畸变(numerical aberration)和结构畸变(structural aberration)两大类,其中染色体的数目畸变又可分为整倍性改变和非整倍性改变。
无论数目畸变,还是结构畸变,其实质是涉及染色体或染色体节段上基因群的增减或位置的转移,使遗传物质发生了改变,结果都可以导致染色体异常综合征,或染色体病。
染色体畸变发生的原因
化学因素
许多化学物质,如一些化学药品、农药、毒物和抗代谢药等,都可以引起染色体畸变。据调查,长期接触苯、甲苯等化学品的人群,出现染色体数目异常和发生染色体断裂的频率远高于一般人群。农药中的除草剂和杀虫的神经剂等都是一些染色体畸变的诱变剂。
药物
某些药物可引起人类染色体畸变或产生畸形胚胎。已有研究证实,环磷酰胺、氮芥、白消安(马利兰)、氮甲蝶啶、阿糖胞苷等抗癌药物可导致染色体畸变;抗疟疾药物采妥英钠可引起人淋巴细胞多倍体细胞数增高。
农药
许多化学合成的农药可以引起人类细胞染色体畸变。如某些有机磷农药可使染色体畸变率增高,如美曲磷酯类农药。
工业毒物
工业毒物如苯、甲苯、铅、砷、二硫化碳、氯丁二烯、氯乙烯单体等,都可以导致染色体畸变。长期接触这些有害毒物的工人,其染色体的畸变率增高。
食品添加剂
某些食品的防腐剂和色素等添加剂中所含的化学物质可以引起人类染色体发生畸变,如硝基咪啶基糖酞胺AF-2、环己基糖精等。
物理因素
细胞到电离辐射后,可引起细胞内染色体发生异常。畸变率随射线剂量的增高而增高;最常见的畸变类型有染色体断裂、缺失、双着丝粒染色体、易位、核内复制、不分离等,这些畸变都可使个体的性状出现异常。
射线的作用包括对体细胞和生殖细胞两方面,如果一次照射大剂量的射线,可在短期内引起造血障碍而死亡。长期接受射线治疗或从事与放射线相关工作的人员,由于微小剂量的射线不断积累,会引起体细胞或生殖细胞染色体畸变。
生物因素
导致染色体畸变的生物因素包括两类:
①由生物体产生的生物类毒素;
②某些生物体如病毒本身可引起染色体畸变;
真菌毒素具有一定的致癌作用,同时也可引起细胞内染色体畸变。如朵色曲霉素、黄曲毒素、棒曲毒素等均可引起染色体畸变;
病毒也可引起宿主细胞染色体畸变,尤其是那些致癌病毒,其原因主要是影响DNA代谢。
当人体感染某些病毒,如风疹病毒、乙肝病毒、麻疹病毒和巨细胞病毒时,就有可能引发染色体的畸变。如果用病毒感染离体培养的细胞将会出现各种类型的染色体异常。
母亲年龄
当母亲年龄增大时,其所生子女的体细胞中某一序号染色体有3条的情况要多于一般人群。
母亲年龄越大(大于35岁),生育Down综合征患儿的危险性就越高。但母亲生育年龄只是环境致畸变因子在体内累积作用的表现形式,这与生殖细胞老化及合子早期所处的宫内环境有关。
一般认为,生殖细胞在母体内停留的时间越长,受到各种因素影响的机会越多,在之后的减数分裂过程中,越容易产生染色体不分离而导致染色体数目异常。
染色体数目异常及其产生机制
人体正常生殖细胞精子和卵子所包含的全部染色体称为一个染色体组。
因此,精子和卵子为单倍体(haploid),以n表示,分别含有22条常染色体和1条性染色体。
受精卵则为二倍体(diploid),以2n表示,包括22对常染色体和1对性染色体。
以人二倍体数目为标准,体细胞的染色体数目(整组或整条)的增加或减少,称为染色体数目畸变。包括整倍性改变和非整倍性改变两种形式。
整倍性改变
如果染色体的数目变化是单倍体n的整倍数,即以n为基数,成倍的增加或减少,则称为整倍性(euploidy)改变。
在2n的基础上,增加一个染色体组n,则染色体数为3n,即三倍体(triploid);若在2n的基础上增加2个n,则为4n,即四倍体(tetraploid)。
三倍体以上的又称为多倍体(polyploid)。如果在2n的基础上减少一个染色体组,则称为单倍体。
非整倍性改变
一个体细胞的染色体数目增加或减少了一条或数条,称非整倍性(aneuploidy)改变,这是临床上最常见的染色体畸变类型。发生非整倍性改变后,会产生亚二倍体(hypodiploid)、超二倍体(hyperdiploid)等。
亚二倍体
当体细胞中染色体数目少了1条或数条时,称为亚二倍体,可写作2n-m(其中m<n)。若果对染色体少了1条(2n-1),细胞染色体数目为45,即构成单体型(monosomy)。临床上常见的单体型有21号、22号和X染色体的单体型。
超二倍体
当体细胞中染色体数目多了一条或数条时,称为超二倍体,可写作2n+m(其中m<n)。在超二倍体的细胞中某一同源染色体的数目不是2条,而是3条,4条……
若某对染色体多了一条(2n+1),细胞内染色体数目为47,即构成该染色体的三体型(trisomy),这是人类染色体数目畸变中最常见、种类最多的一类畸变。由于染色体的增加,特别是较大染色体的增加,将造成基因组的严重失衡而破坏或干扰了胚胎的正常发育,故绝大部分常染色体三体型核型只见于早期流产的胚胎。少数三体型病例可以存活至出生,但多数寿命不长,并伴有各种严重畸形。
三体型以上的非整倍性统称为多体型(polysomy),如四体型、五体型等。多体型常见于性染色体中。
同时存在两种或两种以上核型的细胞系的个体称嵌合体(mosaic)。嵌合体可以是数目异常之间、结构异常之间以及数目和结构异常之间的嵌合。
有时细胞中某些染色体的数目发生了异常,其中有的增加,有的减少,而增加和减少的染色体数目相等,结果是染色体总数不变,还是二倍体数(46条),但不是正常的二倍体核型,则称为假二倍体(pseudodiploid)。
非整倍体的产生原因
多数非整倍体的产生原因是在生殖细胞成熟过程或受精卵早期卵裂中,发生了染色体不分离或染色体丢失。
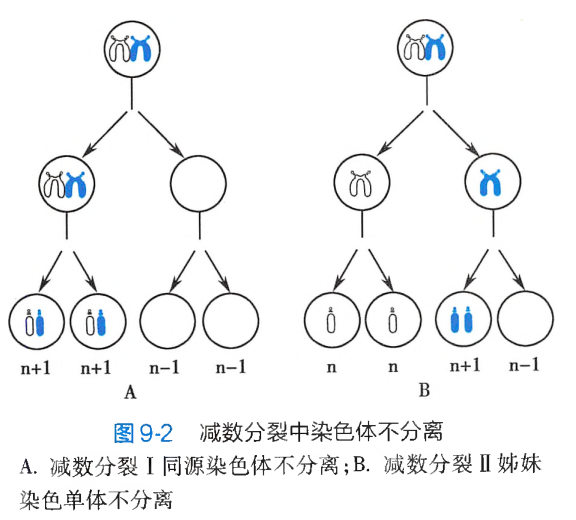
染色体不分离
在细胞分裂进入中、后期时,如果某一对同源染色体或姐妹染色单体彼此没有分离,而是同时进入同一个子细胞,结果所形成的两个子细胞中,一个将因染色体数目增多而成为超二倍体,另一个则因染色体数目减少而成为亚二倍体,这个过程称为染色体不分离(non-disjunction)。
染色体不分离可以发生在细胞的有丝分裂过程中,也可以发生在配子形成时的减数分裂过程。
-
染色体不分离发生在受精卵卵裂早期的有丝分裂过程
-
减数分裂时发生染色体不分离
染色体丢失
染色体丢失(chromosome lose)又称染色体分裂后期滞带(anaphase lag),在细胞有丝分裂过程中,某一染色体未与纺锤丝相连,不能移向两极参与新细胞的形成,或者在移向两极时行动迟缓,滞留在细胞质中,造成该条染色体的丢失而形成亚二倍体。染色体丢失也是嵌合体形成的一种方式。
染色体结构畸变及其产生机制
临床上常见的染色体结构畸变有:缺失、重复、易位、倒位、环状染色体和等臂染色体等。染色体断裂及断裂片段的重接是各种染色体结构畸变产生的基本机制。
缺失
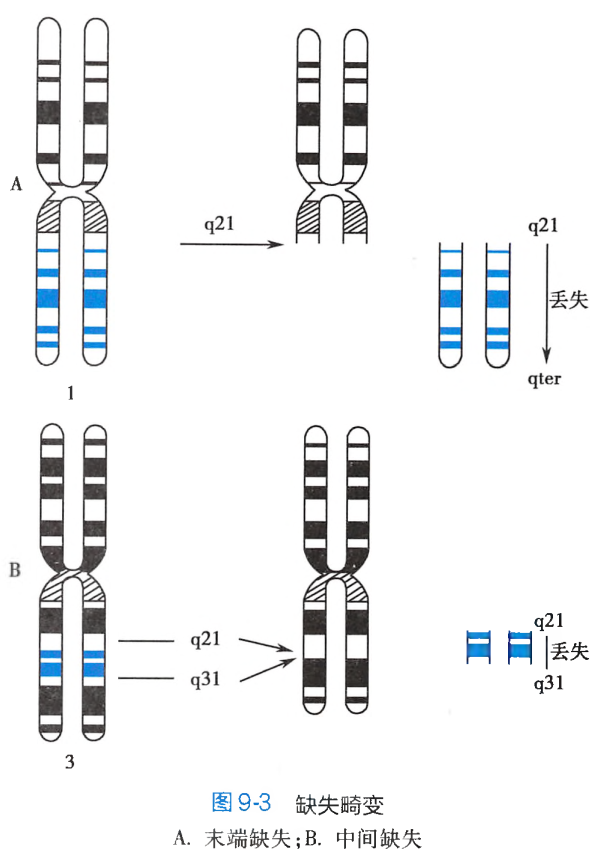
缺失(deletion)是染色体片段的丢失,缺失使位于这个片段的基因也随之发生丢失。按染色体断点的数量和位置可分为末端缺失和中间缺失两类:
-
末端缺失(terminal deletion)指染色体的臂发生断裂后,末发生重接,无着丝粒的片段不能与纺锤丝相连,在细胞分裂后期未能移至两极而丢失。如图9-3A所示,1号染色体长臂的2区1带发生断裂,其远侧段(q21→qter)丢失。这条染色体是由短臂的末端至长臂的2区1带所构成。 -
中间缺失(interstitial deletion)指一条染色体的同一臂上发生了两次断裂,两个断点之间的无着丝粒片段丢失,其余的两个断片重接。如图9-3B所示,3号染色体长臂上的q21和q31发生断裂和重接,这两断点之间的片段丢失。
重复
重复(duplication)是一条染色体上某一片段增加了一份以上的现象,使这些片段的基因多了一份或几份。原因是同源染色体之间的不等交换或姐妹染色单体之间的不等交换以及染色体片段的插入等。
倒位
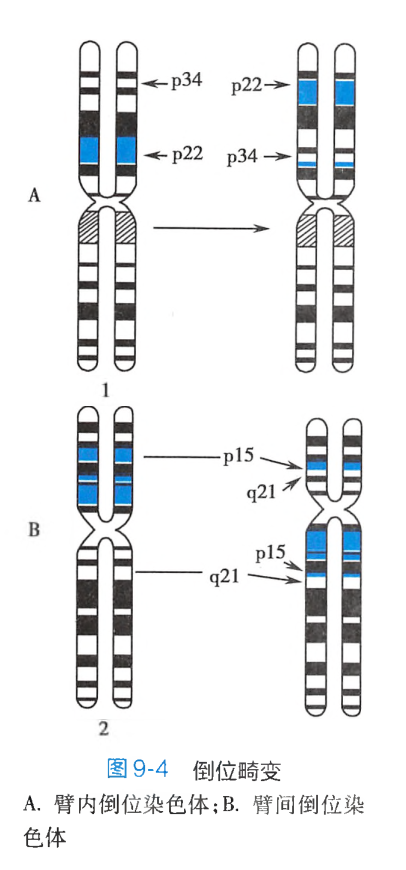
倒位(inversion)是某一染色体发生两次断裂后,两断点之间的片段旋转180°后重接,造成染色体上基因顺序的重排。染色体的倒位可以发生在同一臂(长臂或短臂)内,也可以发生在两臂之间,分别称为臂内倒位和臂间倒位:
-
臂内倒位(paracentric inversion):一条染色体的某一臂上同时发生了两次断裂,两断点之间的片段旋转180°后重接。例如1号染色体p22和p34同时发生了断裂,两断点之间的片段倒转后重接,形成了一条臂内倒位的染色体(图9-4A)。 -
臂间倒位(pericentric inversion):一条染色体的长、短臂各发生了一次断裂,中间断片颠倒后重接,则形成了一条臂间倒位染色体。如2号染色体的p15和q21同时发生了断裂,两断点之间的片段倒转后重接,形成了一条臂间倒位染色体(图9-4B)。
易位
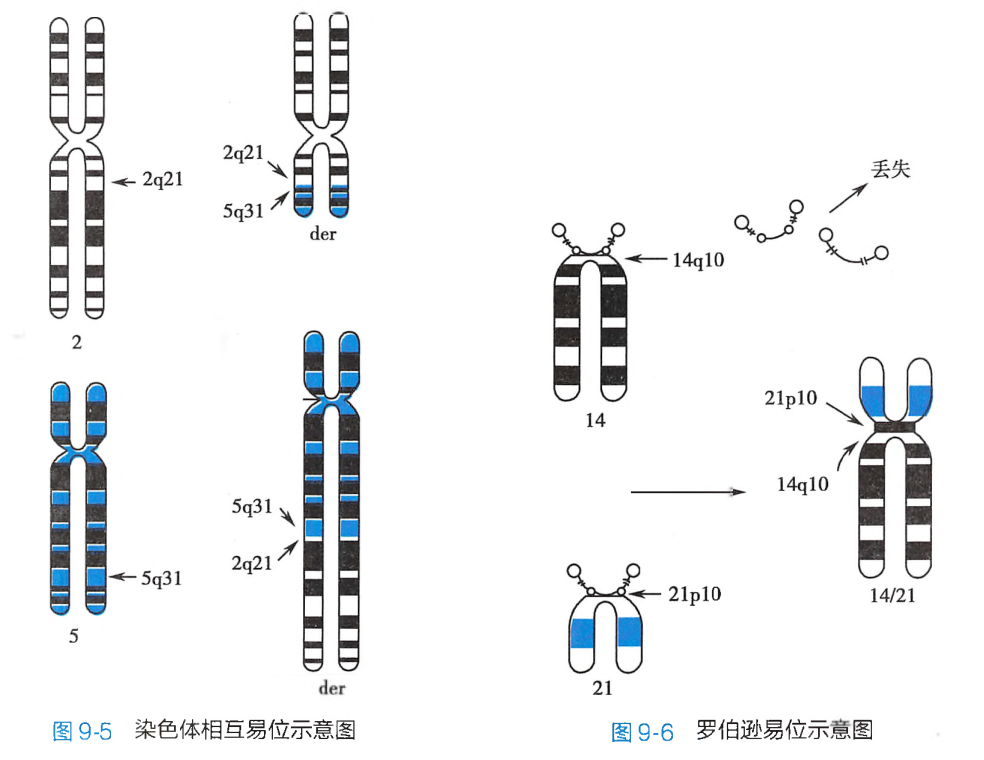
一条染色体的断片移接到另一条非同源染色体的臂上,这种结构畸变称为易位(translocation)。常见的易位方式有相互易位、罗伯逊易位和插入易位等。
相互易位(reciprocal translocation)是两条染色体同时发生断裂,断片交换位置后重接,形成两条衍生染色体(derivative chromosome)。当相互易位仅涉及位置的改变而不造成染色体片段的增减时,称为平衡易位。如2号染色体长臂2区1带和5号染色体长臂3区1带同时发生了断裂,两断片交换位置后重接,形成两条衍生染色体(图9-5)。罗伯逊易位(Robertsonian translocation)又称着丝粒融合(centric fusion)。这是发生于近端着丝粒染色体的一种易位形式。当两个近端着丝粒染色体在着丝粒部位或着丝粒附近部位发生断裂后,两者的长臂在着丝粒处接合在一起,形成一条由两条染色体的长臂构成的衍生染色体;两个短臂则构成一个小染色体,小染色体往往在第一次分裂时丢失(图9-6)。插入易位(insertional translocation),两条非同源染色体同时发生断裂,但只有其中一条染色体的片段插入到另一条染色体的非末端部位。只有发生了三次断裂时,才可能发生插入易位。