
文章链接
Ji Y, Zhou Z, Liu H, Davuluri RV. DNABERT: pre-trained Bidirectional Encoder Representations from Transformers model for DNA-language in genome. Bioinformatics. 2021;37(15):2112-2120. https://doi.org/10.1093/bioinformatics/btab083
简介
DNABERT:提出了一个针对基因组DNA语言的预训练双向编码器Transformers模型
摘要
动机:破译非编码DNA的语言是基因组研究中的一个基本问题。由于多义性和远距离语义关系的存在,基因调控密码高度复杂,而以往的信息学方法在数据稀缺的情况下往往难以捕捉这些特征。
结果:为应对此挑战,我们开发了一种新型的预训练双向编码器表示模型,命名为DNABERT,基于上下游核苷酸上下文来捕获基因组DNA序列的全局和可迁移理解。我们将DNABERT与目前最广泛使用的基因组调控元件预测程序进行了比较,展示了其易用性、准确性和效率。我们证明,单个预训练的transformers模型在使用少量任务特定标记数据进行简单微调后,可以同时在启动子、剪接位点和转录因子结合位点的预测上达到最先进的性能。此外,DNABERT可以直接可视化输入序列中核苷酸级别的重要性和语义关系,从而提供更好的可解释性,并准确识别保守序列基序和功能性遗传变异候选。最后,我们展示了用人类基因组预训练的DNABERT甚至可以直接应用于其他生物体并取得出色的表现。我们预期预训练的DNABERT模型可以微调用于许多其他序列分析任务。
可用性和实现:DNABERT的源代码、预训练模型和微调模型可在GitHub获取(https://github.com/jerryji1993/DNABERT)。
创新点
- 模型创新:
- 首次将
BERT架构应用于DNA序列分析,开发了名为DNABERT的预训练模型 - 创新性地使用
k-mer表示方法对DNA序列进行分词,更好地捕捉局部上下文信息 - 开发了
DNABERT-XL版本以处理超长序列
- 技术优势:
- 通过注意力机制可以全局捕获序列上下文信息
- 采用"自上而下"的方法,先通过自监督预训练理解DNA语言的一般特征,再应用于具体任务
- 在数据稀缺的情况下仍能实现良好性能
- 应用创新:
- 实现"一个模型解决多个任务":同一个预训练模型可以通过简单微调应用于启动子、剪接位点和转录因子结合位点预测等多个任务
- 开发了
DNABERT-viz模块,提供了直观的可视化功能,增强了模型的可解释性 - 能够识别功能性遗传变异
这些创新使得DNABERT在DNA序列分析领域具有重要的理论价值和实际应用价值。
主要内容
读前须知
- 论文解读尽可能的还原原文,若有不恰当之处,还请见谅;
- 排版上,插图会尽量贴近出处,而
补充图表均在文末“支撑性材料”的下载链接中; - 左边👈有目录,可自行跳转至想看的部分;
- 部分专业术语翻译成中文可能不太恰当,此时会用括号标明它的英文原文,如感受野(
Receptive field)。请注意,仅首次出现会标明;
引言
破译DNA中隐藏的指令语言一直是生物研究中的主要目标之一。虽然解释DNA如何转译成蛋白质的遗传密码是通用的,但决定基因何时以及如何表达的调控密码却在不同的细胞类型和生物体中存在差异。相同的顺式调控元件(CREs)在不同的生物学环境中常常具有不同的功能和活性,而远距离分布的多个CREs可能会相互协作,导致在不同功能角色下对替代启动子的依赖性使用。这些观察结果表明序列编码中存在多义性和远距离语义关系,这些都是自然语言的关键特征。先前的语言学研究证实了DNA,尤其是非编码区域,确实表现出与人类语言极大的相似性,从字母和词汇到语法和语音学都有体现。然而,CREs的语义(即功能)如何随不同的上下文(上游和下游核苷酸序列)而变化仍然大部分未知。
近年来,通过成功地将深度学习技术应用于基因组序列数据,已开发出许多计算工具来研究顺式调控景观的各个方面,包括DNA-蛋白质相互作用、染色质可及性、非编码变异等。大多数方法采用基于卷积神经网络(CNN)的架构。其他工具关注DNA的序列特征,试图通过应用基于递归神经网络(RNN)的模型来捕获状态之间的依赖关系,如长短期记忆(LSTM)和门控循环单元(GRU)网络。一些混合方法也被提出来整合这两种模型架构的优点。
为了更好地将DNA建模为一种语言,理想的计算方法应该:
-
全局考虑所有上下文信息以区分多义CREs;
-
发展可转移到各种任务的通用理解;
-
在标记数据有限的情况下具有良好的泛化能力。
然而,CNN和RNN架构都无法满足这些要求。CNN通常无法捕获长程上下文中的语义依赖关系,因为其提取局部特征的能力受限于过滤器大小。RNN模型(LSTM、GRU)虽然能够学习长期依赖关系,但当它顺序处理所有过去的状态并将上下文信息压缩到具有长输入序列的瓶颈时,大大受到梯度消失和低效率问题的影响。此外,大多数现有模型需要大量标记数据,导致在数据稀缺的情况下性能和适用性有限,而在这种情况下,获取高质量的标记数据是昂贵且耗时的。
为了解决上述限制,我们采用了双向编码器表示Transformers(BERT)模型的理念,将其应用于基因组DNA领域,开发了一种称为DNABERT的深度学习方法。DNABERT应用Transformer,这是一种基于注意力的架构,在大多数自然语言处理任务中都取得了最先进的性能。我们证明DNABERT通过以下方式解决了上述挑战:
-
从纯未标记的人类基因组中发展出一般且可迁移的理解,并以"一个模型解决所有问题"的方式通用地解决各种序列相关任务;
-
通过注意力机制全局捕获整个输入序列的上下文信息;
-
在数据稀缺场景中取得出色的性能;
-
无需任何人工指导即可发现DNA序列中的重要子区域和不同顺式元件之间的潜在关系;
-
成功地以跨生物体的方式工作。
由于DNABERT模型的预训练需要大量资源(在8个NVIDIA 2080Ti GPUs上约需25天),作为本研究的主要贡献,我们在GitHub上提供了源代码和预训练模型供未来的学术研究使用。
transformer架构可参考这篇文章:
模型与方法
DNABERT模型
BERT是一种基于transformer的上下文化语言表示模型,在许多自然语言处理(NLP)任务中取得了超人的表现。它引入了预训练和微调的范式,首先从大量未标记的数据中发展通用理解,然后通过最小的架构修改使用特定任务的数据解决各种应用。DNABERT遵循与BERT相同的训练过程。
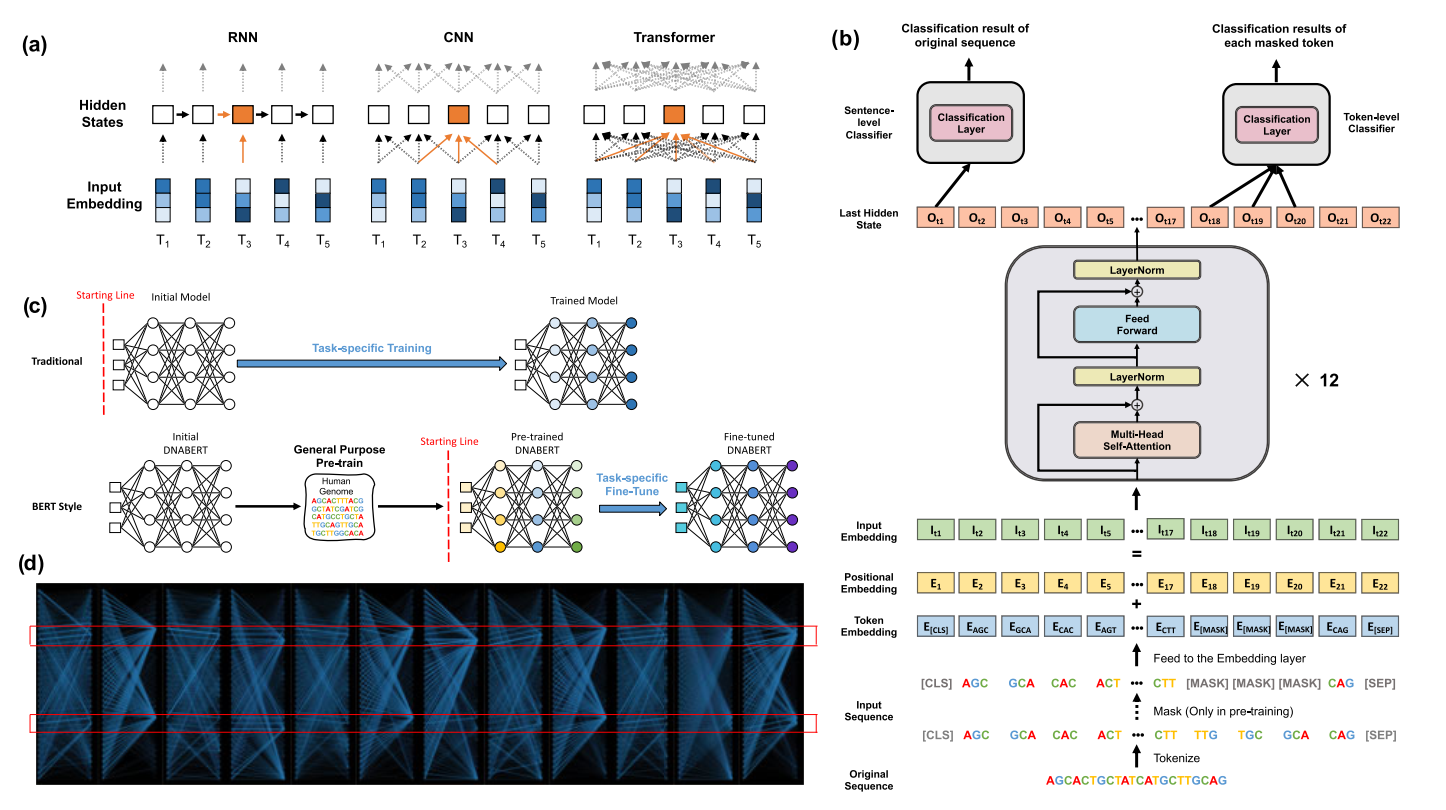
DNABERT首先将一组用k-mer标记表示的序列作为输入(图1b)。每个序列通过将每个标记嵌入到一个数值向量中而表示为一个矩阵M。从形式上讲,DNABERT通过对M执行多头自注意力机制来捕获上下文信息:
其中:
和 是用于线性投影的学习参数。
head通过首先计算每两个标记之间的注意力分数,然后利用它们作为权重来汇总 中的行,从而计算M的下一个隐藏状态。 将h个独立头部的结果与不同的 集合连接起来。整个过程执行L次,L为层数。
与BERT类似,DNABERT也采用预训练-微调方案(图1c)。然而,我们通过删除下一句预测、调整序列长度以及强制模型预测连续k个标记来显著修改了预训练过程,以适应DNA场景。在预训练过程中,DNABERT通过自监督学习掌握DNA的基本语法和语义,基于从人类基因组中通过截断和采样提取的长度为10到510的序列。对于每个序列,我们随机掩盖构成序列15%的k个连续标记区域,让DNABERT基于剩余部分预测被掩盖的序列,确保充足的训练样本。我们使用交叉熵损失预训练DNABERT:
这里, 和 分别是N个类别中每个类别的真实概率和预测概率。预训练的DNABERT模型可以用特定任务的训练数据进行微调,用于各种序列级和标记级预测任务的应用。我们将DNABERT模型在三个特定应用上进行了微调——启动子预测、转录因子结合位点(TFBSs)预测和剪接位点预测——并将训练好的模型与当前最先进的工具进行了基准比较。
DNABERT模型的训练
标记化
与将每个碱基视为单个标记不同,我们使用k-mer表示法对DNA序列进行标记化,这是一种在分析DNA序列时被广泛使用的方法。k-mer表示通过将每个脱氧核苷酸碱基与其后续碱基连接起来,为其整合了更丰富的上下文信息。它们的连接被称为一个k-mer。例如,一个DNA序列"ATGGCT"可以被标记化为四个3-mer:{ATG, TGG, GGC, GCT}或两个5-mer:{ATGGC, TGGCT}。由于不同的k会导致DNA序列的不同标记化。在我们的实验中,我们分别将k设置为3、4、5和6,并训练了4个不同的模型:DNABERT-3、DNABERT-4、DNABERT-5、DNABERT-6。对于DNABERT-k,它的词汇表由所有k-mer的排列组合以及5个特殊标记组成:[CLS]代表分类标记;[PAD]代表填充标记,[UNK]代表未知标记,[SEP]代表分隔标记,[MASK]代表掩码标记。因此,在DNABERT-k的词汇表中有4k+5个标记。
预训练
跟随之前的工作,DNABERT以最大长度为512的序列作为输入。如图1b所示,对于一个DNA序列,我们首先将其标记化为一系列k-mers,并在其开始处添加一个特殊的[CLS]标记(代表整个序列),以及在末尾添加一个特殊的[SEP]标记(表示序列结束)。在预训练步骤中,我们掩盖了某些k-mers的连续k长度跨度(总计输入序列的15%),考虑到一个标记可能会从紧邻的k-mers中被轻易推断出来,而在微调时,我们跳过掩码步骤,直接将标记化的序列输入到嵌入层。
我们通过两种方法从人类基因组生成训练数据:直接非重叠分割和随机采样,序列长度在5到510之间。我们以2000的批量大小对DNABERT进行了120k步的预训练。在前100k步中,我们在每个序列中掩盖了15%的k-mers。在最后20k步中,我们将掩码率提高到20%。学习率在前10k步中从0线性增加(即预热)到,然后在200k步后线性降至0(补充图S1)。我们在120k步后停止训练过程,因为我们发现损失曲线出现了平稳的迹象。
我们使用与BERT base相同的模型架构,它由12个Transformer层组成,每层有768个隐藏单元和12个注意力头,并在预训练期间对所有四个DNABERT模型使用相同的参数设置。我们在配备8个Nvidia2080Ti GPUs的机器上使用混合精度浮点运算训练每个DNABERT模型。
微调
对于每个下游应用,我们从预训练参数开始,使用特定任务的数据对DNABERT进行微调。我们在所有应用中都使用了相同的训练技巧,即学习率首先线性预热到峰值,然后线性衰减到接近0。我们使用固定权重衰减的AdamW作为优化器,并对输出层采用了dropout。我们将训练数据分为训练集和开发集用于超参数调优。对于不同k值的DNABERT,我们稍微调整了峰值学习率。详细的超参数设置列在补充表S5中。
对于长度超过512的序列,我们将它们分割成片段并连接它们的表示作为最终表示。这使得DNABERT能够处理超长序列(DNABERT-XL)。k = 3、4、5、6的DNABERT都取得了非常相似的性能,只有轻微波动。在所有实验中,我们报告kmer=6的结果,因为它取得了最佳性能。
结果
DNABERT-Prom能够有效预测近端和核心启动子区域
预测基因启动子是生物信息学中最具挑战性的问题之一。我们首先评估了预训练模型在识别近端启动子区域方面的表现。为了公平地与具有不同序列长度设置的现有工具进行比较,我们使用来自真核生物启动子数据库(EPDnew)的长度为10000bp的人类TATA和非TATA启动子,对两个模型进行了微调,分别命名为DNABERT-Prom-300和DNABERT-Prom-scan。我们使用TSS周围从-249到50bp的序列作为正例,随机选择的300bp长度含TATA序列作为TATA负例,以及二核苷酸打乱的序列作为非TATA负例,将DNABERT-Prom-300与DeePromoter进行了比较。我们使用基于10000bp长序列的滑动窗口扫描,将DNABERT-Prom-scan与当前可用的方法进行了比较,包括最新的最先进方法PromID、FPROM和我们之前的软件FirstEF。为了适当地在相同设置下与PromID进行基准比较,我们使用了1001bp长的扫描,这超出了传统BERT模型的长度容量。因此,我们专门为此任务开发了DNABERT-XL。我们使用与PromID相同的评估标准,通过扫描序列并将预测与已知TSS的-500到+500bp重叠。与已知TSS的-500到+500bp重叠≥50%的1001bp序列被视为阳性,其余的被视为阴性。对于PromID和FPROM,测试集被直接输入进行评估。相比之下,FirstEF首先生成全基因组预测,然后将其与阳性序列对齐。
DNABERT-Prom在不同设置下通过显著提高的准确性指标超越了所有其他模型(图2)。具体而言,对于prom-300设置下的TATA启动子,DNABERT-Prom-300在准确率和MCC指标上分别超过DeePromoter 0.335和0.554(图2a)。同样,我们在非TATA和组合情况下也观察到DNABERT-Prom的性能显著提高(补充图S2)。同时,prom-scan设置本质上更加困难,因为类别高度不平衡,所以所有测试的基准模型表现都很差。在基准模型中,FirstEF取得了最好的性能,在TATA、非TATA和组合数据集的F1得分分别为0.277、0.377和0.331(图2b)。然而,DNABERT-Prom-scan取得的F1得分和MCC远超FirstEF。
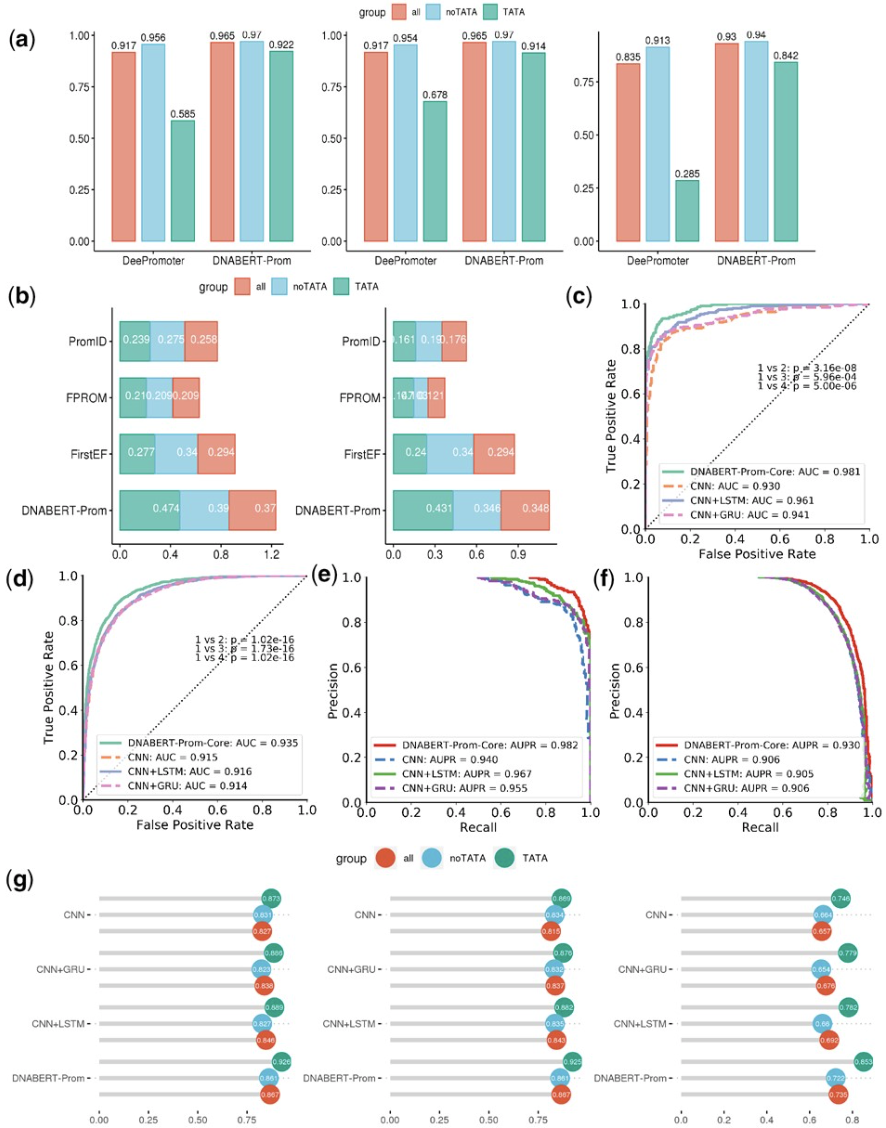
接下来,我们评估了我们的模型在核心启动子上的预测性能,这是一个由于序列上下文大小减少而更具挑战性的问题。我们使用了以TSS为中心的Prom-300数据中的70bp,并与CNN、CNN+LSTM和CNN+GRU进行了比较。DNABERT-Prom-core在不同数据集上明显优于所有三个基准模型(图2c-g),清楚地表明DNABERT可以被可靠地微调,仅依靠TSS区域附近的序列模式就能准确预测长的近端启动子和较短的核心启动子。为了进一步证明DNABERT-XL的有效性,我们还在301bp长序列和2001bp长序列上进行了实验。实验表明,该模型在预测2001 bp长序列时取得了更好的性能(补充表S7)。
DNABERT-TF能准确识别转录因子结合位点
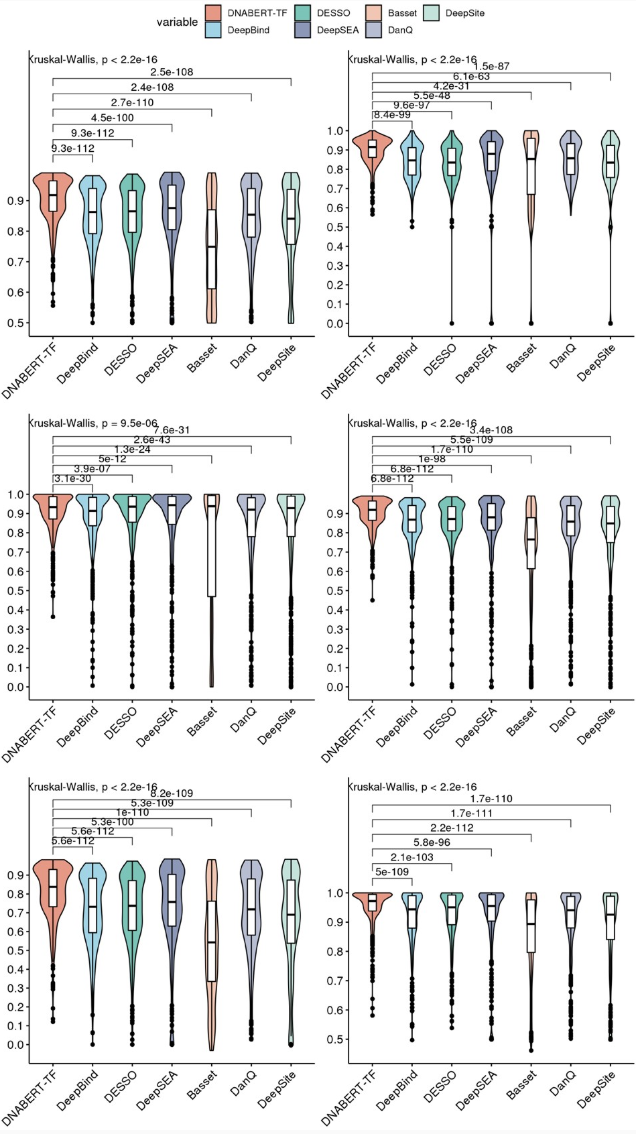
第二代测序技术以前所未有的方式促进了基因组调控区域的全基因组鉴定,揭示了基因调控的复杂性。在分析体内基因组范围内结合相互作用数据时,一个重要步骤是预测目标顺式调控区域中的转录因子结合位点(TFBS)并整理得到的转录因子结合谱。因此,我们使用来自ENCODE数据库的690个TF ChIP-seq统一峰值谱来微调DNABERT-TF模型,以预测ChIP-seq富集区域中的TFBS,并与广泛使用的和之前发表的TFBS预测工具进行了比较,包括DeepBind、DeepSEA、Basset、DeepSite、DanQ和DESSO。
DNABERT-TF是唯一一个平均和中位数准确率和F1值都超过0.9的方法(图3,0.918和0.919),大大超过第二好的竞争者(DeepSEA,Wilcoxon单侧符号秩检验,n=690,校正后P=和,对于平均值)。其他工具在某些实验中产生了许多假阳性(FP)和假阴性(FN)预测,导致在比较平均值时表现更不令人满意,这是由于分布的偏斜性。几个工具在使用高质量数据的实验中,在找到真阴性(TN)方面与DNABERT表现相当,但在低质量实验数据的预测中表现较差。相比之下,即使在低质量数据上,DNABERT也实现了显著高于其他工具的召回率(图3,中左)。同时,无论实验质量如何,DNABERT-TF产生的假阳性预测都比任何其他模型少得多(图3,右上)。这些结果在使用有限数量峰值的ChIP-seq谱的基准测试中得到进一步支持,其中DNABERT-TF始终优于其他方法(见补充图S3)。
为了评估我们的方法是否能有效区分多义的顺式调控元件,我们关注了p53家族蛋白(它们识别相同的基序),并研究了TAp73-alpha和TAp73-beta亚型之间结合特异性的上下文差异。我们将来自GEO数据集GSE15780的p53、TAp73-alpha和TAp73-beta ChIP-seq峰值与我们的P53Scan程序预测的结合位点重叠,并使用得到的ChIP-seq特征化BS(35bp)来微调我们的模型。DNABERT-TF在个别TF的二元分类上实现了接近完美的表现(0.99)(见补充表S2)。使用具有更宽上下文(500bp)的输入序列,DNABERT-TF能有效区分两种TAp73亚型,准确率达到0.828(见补充表S2)。总之,DNABERT-TF可以根据不同的上下文窗口准确识别甚至非常相似的TFBS。
DNABERT-viz 实现重要区域、上下文和序列基序的可视化
为了克服常见的"黑箱"问题,深度学习模型需要在与传统方法相比表现出色的同时保持可解释性。因此,为了总结和理解微调后的DNABERT模型基于哪些重要序列特征做出分类决策,我们开发了DNABERT-viz模块,用于直接可视化对模型决策有贡献的重要区域。我们证明,由于注意力机制的存在,DNABERT天生适合在DNA序列中找到重要模式并理解它们在上下文中的关系,从而确保了模型的可解释性。
图4a显示了三个TAp73-beta响应元件的学习到的注意力图,其中DNABERT-viz以无监督的方式准确确定了P53Scan预测的TFBS的位置和得分。然后,我们汇总了所有热图,在Prom-300和ENCODE 690 TF的测试集上产生注意力景观。对于TATA启动子,DNABERT始终在TSS上游-20到-30bp区域(TATA框所在位置)表现出高度注意力,而对于大多数非TATA启动子,则观察到更分散的注意力模式(图4)。这种模式也在TF-690数据集中看到,每个峰值都显示出一组不同的高注意力区域,其中大多数分散在峰值中心周围(补充图S4)。我们特别关注了个别ChIP-seq实验的例子,以更好地理解注意力模式。大多数高质量实验都显示在ChIP-seq峰值中心或TFBS区域周围的注意力富集(图4和补充图S5)。相比之下,低质量实验倾向于具有分散的注意力,没有明显可观察到的模式,除了仅在序列开始处的高注意力,这可能是由于模型偏差造成的(图4d)。
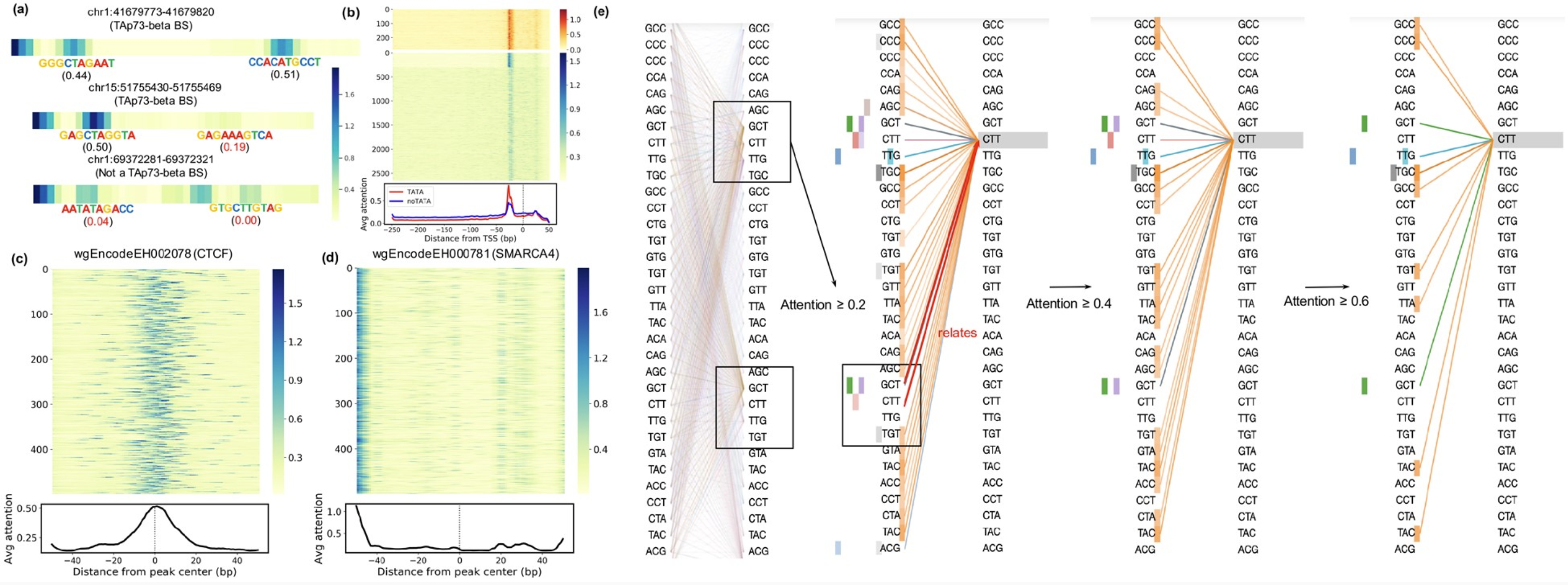
接下来,我们扩展了DNABERT-viz,使其能够直接可视化任何输入序列内的上下文关系(图4e)。例如,最左边的图显示了p53数据集中一个输入序列的全局自注意力模式,其中来自大多数k-mer标记的各个注意力在所有头部中都正确地集中在二聚体BS的两个中心。通过观察哪些标记特别关注该位点,我们可以进一步推断BS与输入序列其他区域之间的相互依赖关系(图4e,右)。在注意力头部中,橙色的头部明显发现了上下文中隐藏的语义关系,因为它广泛突出了对这个重要标记CTT的注意力有贡献的各个短区域。此外,三个头部(绿色、紫色和粉色)成功地将这个标记与二聚体BS的下游一半相关联,展示了对输入序列的上下文理解。
为了在许多输入序列中提取保守的基序模式,我们应用DNABERT-viz来寻找连续的高注意力区域,并通过超几何检验进行过滤(见补充方法)。然后将得到的显著基序实例对齐并合并,生成位置权重矩阵(PWMs)。通过在ENCODE 690数据集中发现的基序上应用TOMTOM程序并与JASPAR 2018数据库比较,我们发现发现的1999个基序中有1595个成功对齐到已验证的基序(补充图S6,q值<0.01)。通过与已记录基序的强烈相似性,说明识别的基序总体上质量很高(补充图S7)。
最后,我们应用DNABERT-viz来理解区分TAp73-alpha和beta亚型结合位点的重要因素。注意力景观确实显示出两种亚型之间差异富集的许多短区域,其中alpha在中心具有更集中的高注意力,而beta则更分散在上下文中(补充图S8)。提取的许多强基序模式都没有对齐到JASPAR数据库,除了一些突出未知关系的模式(补充图S9)。重要的是,c-Fos、c-Jun和TAp73-alpha/beta亚型之间的差异性串扰对细胞凋亡平衡有贡献,而DNABERT-viz成功捕捉到了这种关系。总之,DNABERT可以以更直接的方式获得与基于CNN的模型相当的可解释性,同时在预测性能上大大超越它们。
DNABERT-Splice准确识别经典和非经典剪接位点
预测剪接位点对于揭示基因结构和理解选择性剪接机制至关重要。然而,既存在含有GT-AG的非剪接位点序列,又存在不含这些二核苷酸的非经典剪接位点,这给准确识别带来了困难。最近,SpliceFinder通过递归纳入先前误分类的假阳性序列来重建数据集,成功解决了这个问题。为了与SpliceFinder在相同基准数据上的性能进行比较,我们迭代重建了包含供体、受体和非剪接位点类别的相同数据集。我们还与多个基线模型进行了比较分析。
正如预期的那样,由于任务过于简单化,所有模型在初始数据集上的表现都很好,尽管DNABERT-Splice仍然取得了最好的成绩(补充表S3)。然后,我们使用包含"对抗样本"的重建数据集将DNABERT-Splice与所有基线进行比较(图5a)。这一次,基线模型的预测性能大幅下降,而DNABERT-Splice仍然实现了0.923的最佳准确率、0.919的F1值和0.871的MCC,其AUROC和AUPRC显著优于其他模型(图5b和c),这也得到了Mcnemar精确检验的支持(补充图S10和S11)。此外,当在包含我们迭代训练过程中保留的滑动窗口扫描的独立测试集上进行预测时,DNABERT-Splice再次优于所有模型(补充表S4)。
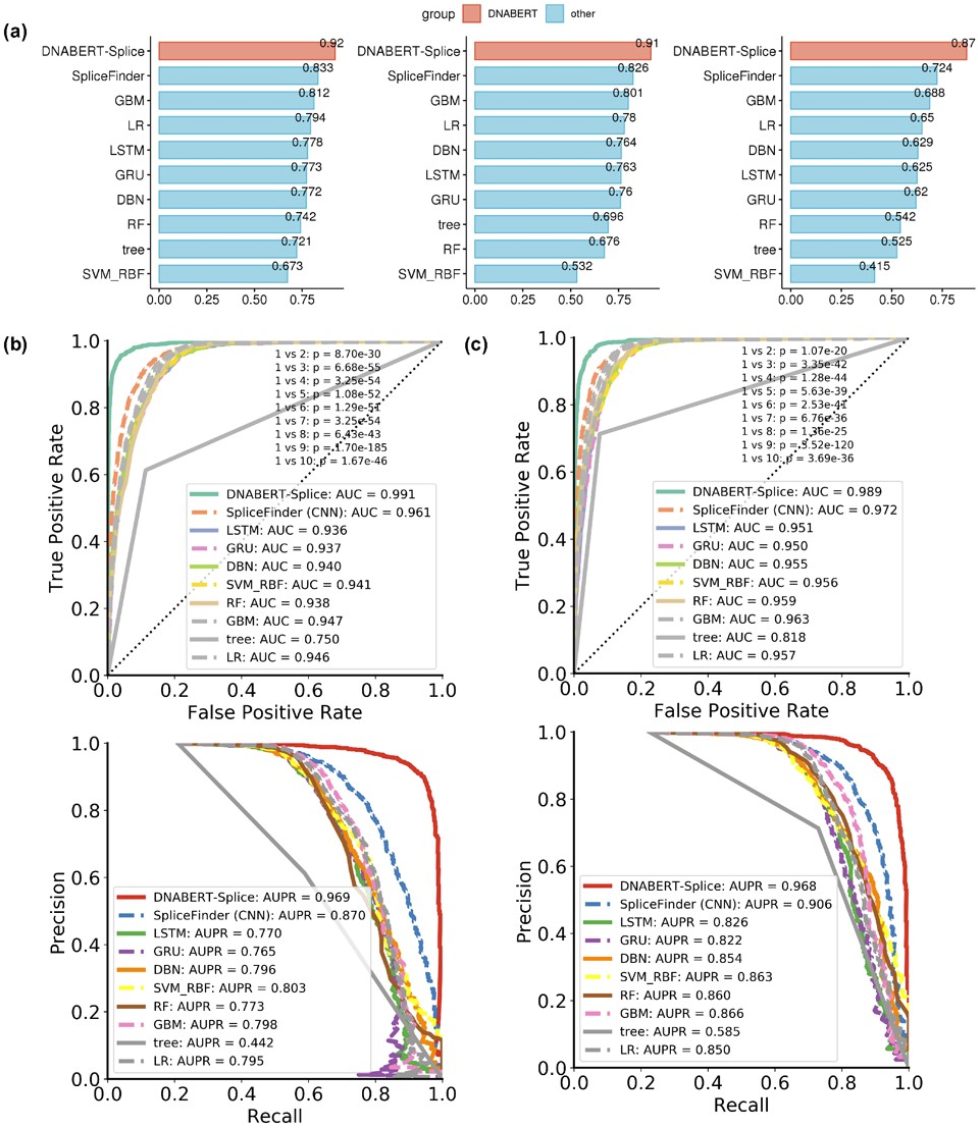
我们还检查了注意力景观,以阐明模型如何做出分类决策(补充图S12)。令人惊讶的是,DNABERT-Splice在内含子区域(供体下游和受体上游)表现出全局一致的高注意力,突显了作为剪接CREs的各种内含子剪接增强子(ISEs)和抑制子(ISSs)的存在和功能重要性。
预训练显著提升性能并可推广到其他生物体
最后,我们基于性能提升和可推广性研究了预训练的重要性。当在相同超参数下比较预训练的DNABERT-prom-300与随机初始化的训练损失时,预训练的DNABERT收敛到明显更低的损失,这表明没有预训练的随机初始化模型很快就陷入局部最小值,因为预训练通过捕获远距离上下文信息确保了对DNA逻辑的初步理解(图6d)。同样,随机初始化的DNABERT-prom-core模型要么完全无法训练,要么表现出次优性能。
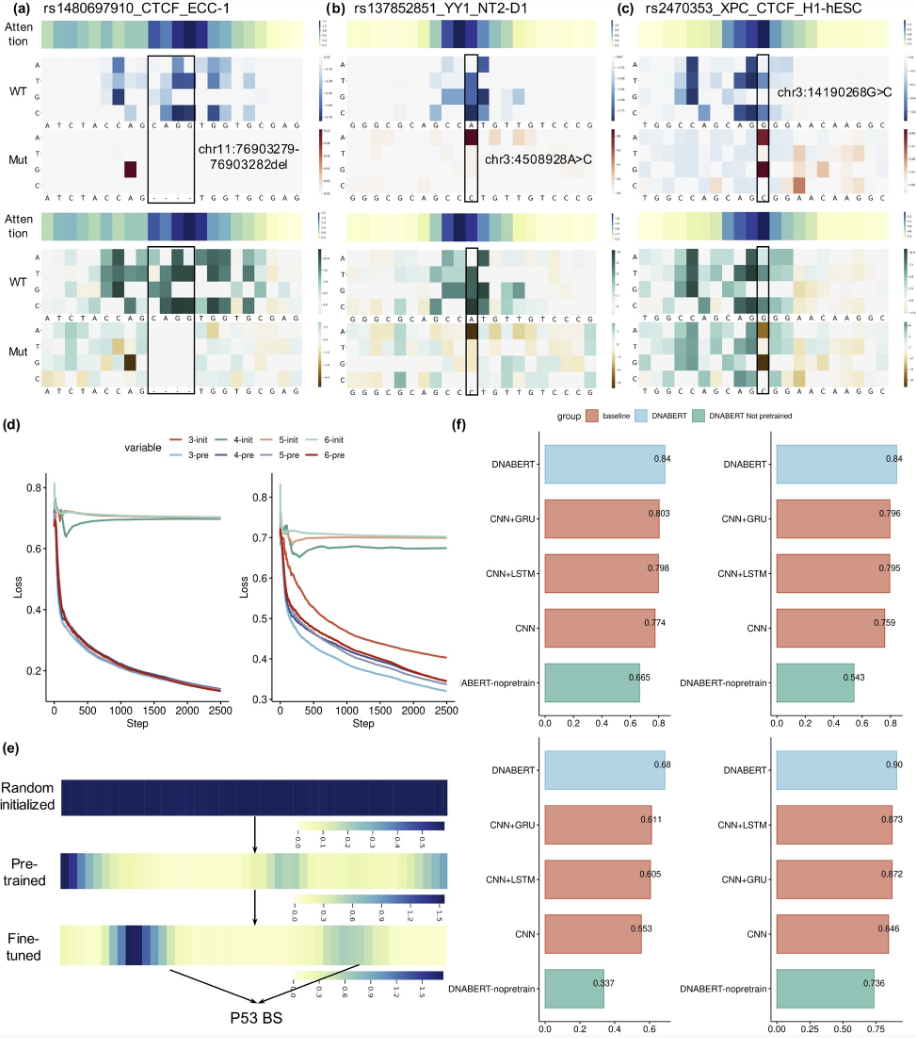
对注意力图的检查揭示了对输入序列的逐步理解(图6e)。由于对不同生物体分别预训练DNABERT既耗时又需要大量资源,我们还评估了用人类基因组预训练的DNABERT是否也可以应用于其他哺乳动物生物体。具体来说,我们用78个小鼠ENCODE ChIP-seq数据集对用人类基因组预训练的DNABERT进行微调,并与CNN、CNN+LSTM、CNN+GRU和随机初始化的DNABERT进行比较。预训练的DNABERT显著优于所有基线模型(图6f),表明即使在不同基因组间DNABERT也具有稳健性和适用性。
众所周知,虽然人类和小鼠基因组的蛋白质编码区域约有85%的同源性,但非编码区域仅显示约50%的全局相似性。由于TFBS主要位于非编码区域,DNABERT模型成功地将学习到的信息从一个基因组转移到一个相似度低得多的基因组,对这些差异具有很高的容忍度。这表明该模型正确捕获了不同生物体DNA序列中共同的深层语义。上述评估证明了预训练的必要性,并保证了预训练模型在不同生物体的众多序列预测任务中的高效应用的可扩展性。
讨论
基于Transformers的模型在各种自然语言处理任务、大规模电子健康记录备注和生物医学文档的生物医学和临床实体提取方面都取得了最先进的性能。之前的研究已经将Transformers应用于蛋白质序列和原核生物基因组。在这里,我们展示了DNABERT通过大大超越现有工具,在各种下游DNA序列预测任务中实现了卓越的性能。通过对输入序列创新性的全局上下文嵌入,DNABERT采用"自上而下"的方法来解决序列特异性预测问题,首先通过自监督预训练发展对DNA语言的普遍理解,然后将其应用于特定任务,这与使用特定任务数据的传统"自下而上"方法形成对比。
DNABERT的这些特点确保它能更有效地从DNA上下文中学习,具有适应多种情况的巨大灵活性,并且在有限数据条件下性能得到提升。特别是,我们还观察到预训练的DNABERT在不同生物体之间具有很强的泛化能力,这确保了我们的方法无需单独预训练就能广泛应用。
作为本研究的一部分发布的预训练DNABERT模型可以用于其他序列预测任务,例如,从ATAC-seq和DAP-seq确定CREs和增强子区域。此外,由于RNA序列与DNA序列仅相差一个碱基(胸腺嘧啶变成尿嘧啶),而语法和语义基本保持不变,我们提出的方法也可以应用于交联和免疫沉淀(CLIP-seq)数据,用于预测RNA结合蛋白(RBPs)的结合偏好。尽管对DNA的直接机器翻译尚不可能,但DNABERT的成功开发为这种可能性提供了启示。
作为一个成功的语言模型,DNABERT正确捕获了DNA序列中隐藏的语法、语法规则和语义,一旦标记级标签可用,它应该在序列到序列(Seq2seq)翻译任务上表现同样出色。同时,DNA和人类语言在文本之外的其他相似方面(如选择性剪接和标点符号)突显了需要结合不同层次的数据来更恰当地破译DNA语言。总之,我们预期DNABERT通过将先进的语言建模视角带入基因调控分析,可以为生物信息学界带来新的进展和见解。
支撑性材料
本文的支撑性材料可在这里获取。
参考文献
-
Andersson, R., Sandelin, A. (2020). Determinants of enhancer and promoter activities of regulatory elements.
-
Nirenberg, M., Leder, P., Bernfield, M., Brimacombe, R., Trupin, J., Rottman, F., O’neal, C. (1965). RNA codewords and protein synthesis, VII. On the general nature of the RNA code.
-
Davuluri, R.V., Suzuki, Y., Sugano, S., Plass, C., Huang, T.H.-M. (2008). The functional consequences of alternative promoter use in mammalian genomes.
-
Gibcus, J.H., Dekker, J. (2012). The context of gene regulation.
-
Ji, Y., Mishra, R.K., Claude, E., Moore, J.E., Lobos, C.M., Hoff, A.M., et al. (2020). Promoter-proximal pausing coordinates tissue-specific timing of gene expression.
-
Vitting-Seerup, K., Sandelin, A. (2017). The landscape of isoform switches in human cancers.
-
Brendel, V., Busse, H.G. (1984). Genome structure described by formal languages.
-
Head, T. (1987). Formal language theory and DNA: An analysis of the generative capacity of specific recombinant behaviors.
-
Ji, S. (1999). The linguistics of DNA: Words, sentences, grammar, phonetics, and semantics.
-
Mantegna, R.N., Buldyrev, S.V., Goldberger, A.L., Havlin, S., Peng, C.-K., Simons, M., Stanley, H.E. (1994). Linguistic features of noncoding DNA sequences.
-
Searls, D.B. (1992). The linguistics of DNA.
-
Searls, D.B. (2002). The language of genes.
-
Alipanahi, B., Delong, A., Weirauch, M.T., Frey, B.J. (2015). Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning.
-
Kelley, D.R., Snoek, J., Rinn, J.L. (2016). Basset: Learning the regulatory code of the accessible genome with deep convolutional neural networks.
-
Zhou, J., Troyanskaya, O.G. (2015). Predicting effects of noncoding variants with deep learning-based sequence model.
-
Zou, J., Huss, M., Abid, A., Mohammadi, P., Torkamani, A., Telenti, A. (2019). A primer on deep learning in genomics.
-
Hochreiter, S., Schmidhuber, J. (1997). Long short-term memory.
-
Cho, K., Van Merriënboer, B., Gulcehre, C., Bahdanau, D., Bougares, F., Schwenk, H., Bengio, Y. (2014). Learning phrase representations using RNN encoder-decoder for statistical machine translation.
-
Hassanzadeh, H.R., Wang, M.D. (2016). DeeperBind: Enhancing prediction of sequence specificities of DNA binding proteins.
-
Quang, D., Xie, X. (2016). DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences.
-
Shen, Z., Bao, W., Huang, D.-S. (2018). Recurrent neural network for predicting transcription factor binding sites.
-
Bengio, Y., Courville, A., Vincent, P. (2013). Representation learning: A review and new perspectives.
-
LeCun, Y., Bengio, Y., Hinton, G. (2015). Deep learning.
-
Devlin, J., Chang, M.-W., Lee, K., Toutanova, K. (2018). BERT: Pre-training of deep bidirectional transformers for language understanding. https://arxiv.org/abs/1810.04805
-
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A.N., et al. (2017). Attention is all you need.
-
Liu, Y., Ott, M., Goyal, N., Du, J., Joshi, M., Chen, D., et al. (2019). RoBERTa: A robustly optimized BERT pretraining approach.
-
Yang, Z., Dai, Z., Yang, Y., Carbonell, J., Salakhutdinov, R.R., Le, Q.V. (2019). XLNet: Generalized autoregressive pretraining for language understanding.
-
Li, F., Chen, Y., Zhang, Z., Ouyang, J., Wang, Y., Meng, S., et al. (2019). Discriminating clinical phases of recovery from major depressive disorder using the dynamics of facial expression.
-
Lee, J., Yoon, W., Kim, S., Kim, D., Kim, S., So, C.H., Kang, J. (2020). BioBERT: a pre-trained biomedical language representation model for biomedical text mining.
-
Clauwaert, J., Waegeman, W. (2020). Novel transformer networks for improved sequence labeling in genomics.
-
Min, X., Zeng, W., Chen, S., Chen, N., Chen, T., Jiang, R. (2019). Predicting enhancers with deep convolutional neural networks.
-
Gupta, S., Stamatoyannopoulos, J.A., Bailey, T.L., Noble, W.S. (2007). Quantifying similarity between motifs.
-
Koeppel, M., van Heeringen, S.J., Kramer, D., Smeenk, L., Janssen-Megens, E., Hartmann, M., et al. (2011). Crosstalk between c-Jun and TAp73α/β contributes to the apoptosis-survival balance.
-
Wang, Z., Burge, C.B. (2008). Splicing regulation: from a parts list of regulatory elements to an integrated splicing code.
-
Buenrostro, J.D., Giresi, P.G., Zaba, L.C., Chang, H.Y., Greenleaf, W.J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position.
-
Bartlett, A., O’malley, R.C., Huang, S.C., Galli, M., Nery, J.R., Gallavotti, A., Ecker, J.R. (2017). Mapping genome-wide transcription-factor binding sites using DAP-seq.
-
Gerstberger, S., Hafner, M., Tuschl, T. (2014). A census of human RNA-binding proteins.
-
Mouse Genome Sequencing Consortium (2002). Initial sequencing and comparative analysis of the mouse genome.
-
Mouse ENCODE Consortium (2012). An encyclopedia of mouse DNA elements (Mouse ENCODE).
