
文章链接
Cui, H., Wang, C., Maan, H., Pang, K., Luo, F., Duan, N., & Wang, B. (2024). scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nature Methods, 21,1470-1480. https://doi.org/10.1038/s41592-024-02201-0
简介
scGPT:提出了一个利用生成式AI构建的单细胞多组学基础模型。
摘要
生成式预训练模型在语言和计算机视觉等多个领域取得了显著成功。具体而言,大规模多样化数据集与预训练transformer的结合已经成为开发基础模型的一种很有前景的方法。基于语言和细胞生物学之间的相似性(文本由词语组成;类似地,细胞由基因定义),我们的研究探索了基础模型在推进细胞生物学和遗传学研究方面的应用潜力。利用蓬勃发展的单细胞测序数据,我们基于生成式预训练transformer在超过3300万个细胞的数据库上构建了一个单细胞生物学基础模型scGPT。我们的研究表明,scGPT能有效提取关于基因和细胞的关键生物学见解。通过进一步采用迁移学习,scGPT可以针对各种下游应用进行优化以实现卓越性能。这包括细胞类型注释、多批次整合、多组学整合、扰动响应预测和基因网络推断等任务。
创新点
- 首个单细胞组学领域的
基础模型
- 首次将大规模预训练
transformer应用于单细胞组学数据分析 - 在超过3300万个细胞的数据集上进行预训练
- 可以通过微调适应多种下游任务
- 创新的模型架构
- 设计了特殊的
注意力掩码机制,可以处理非序列的基因表达数据 - 同时学习基因和细胞的嵌入表示
- 支持通过
基因提示和细胞提示进行生成式预测
- 多功能性和通用性
- 在多个下游任务中实现了最先进的性能,包括:
- 细胞类型注释
- 基因扰动响应预测
- 多批次整合
- 多组学整合
- 基因网络推断
- 展现了强大的
零样本和少样本迁移学习能力
- 可解释性
- 通过注意力机制学习
基因-基因相互作用 - 可以揭示特定细胞状态下的基因网络激活模式
- 发现的基因调控关系与已知生物学知识高度吻合
主要内容
读前须知
- 论文解读尽可能的还原原文,若有不恰当之处,还请见谅;
- 排版上,插图会尽量贴近出处,而
补充图表均在文末“补充信息”的下载链接中; - 左边👈有目录,可自行跳转至想看的部分;
- 部分专业术语翻译成中文可能不太恰当,此时会用括号标明它的英文原文,如感受野(
Receptive field)。请注意,仅首次出现会标明;
引言
单细胞RNA测序(scRNA-seq)通过实现对不同细胞类型的精细表征并推进我们对疾病发病机制的理解,为细胞异质性探索、谱系追踪、致病机制阐明以及最终的个性化治疗策略铺平了道路。scRNA-seq的广泛应用产生了诸如人类细胞图谱等全面的数据图谱,现在已经包含数千万个细胞。测序技术的最新进展促进了数据模态的多样性,并将我们的认知从基因组学扩展到表观遗传学、转录组学和蛋白质组学,从而提供多模态的见解。这些突破也带来了新的研究问题,如参考映射、扰动预测和多组学整合。开发能够有效利用、增强和适应测序数据快速扩张的方法是至关重要的。
参考映射(Reference Mapping):将测序数据比对到已知的参考基因组序列上的过程;
扰动预测(Perturbation Prediction):预测对生物系统施加干扰后可能产生的变化或响应;
多组学整合(Multi-omics Integration):将不同层面的组学数据(如基因组学、转录组学、蛋白组学等)进行综合分析,以获得系统性的认识。
解决这一挑战的一个有前景的方法是生成式预训练基础模型。基础模型通常基于self-attention transformer架构构建,因其在学习数据表征方面的有效性而被广泛采用,这类模型在大规模、多样化数据集上进行预训练,可以轻松适应各种下游任务。此类模型最近在多个领域取得了空前的成功,典型例子包括计算机视觉和自然语言生成领域的DALL-E 2和GPT-4,以及最近用于生物应用的Enformer。
生成式预训练模型(Generative Pre-trained Models):先在大规模数据上进行无监督预训练,再针对特定任务微调的人工智能模型,能够生成文本、图像等内容。典型代表如GPT、BERT等。这类模型通过"预训练-微调"的范式,可以学习到通用的知识表示,并应用于各种下游任务。
更有趣的是,这些生成式预训练模型始终优于从头训练的特定任务模型。这表明这些模型在相应领域获得了与任务无关的知识理解,启发我们探索其在单细胞组学研究中的应用。然而,当前单细胞研究中的机器学习方法较为分散,具体模型仅专注于特定的分析任务。因此,每项研究使用的数据集在广度和规模上往往受限。为了应对这一局限性,需要一个在大规模数据上预训练的基础模型,能够理解不同组织中基因之间的复杂相互作用。
为了增强单细胞测序数据的建模,我们从自然语言生成(NLG)中的自监督预训练工作流程获得灵感,其中self-attention transformer在建模输入词语标记方面表现出了强大的能力。正如文本由词语构成,细胞可以通过基因及其编码的蛋白质产物来表征。通过同时学习基因和细胞嵌入,我们可以更好地理解细胞特征。此外,transformer输入标记的灵活性使其能够轻松整合额外的特征和元信息。最近在Geneformer中也探索了这一方向,其中基于transformer的编码器通过按表达水平排序的基因进行训练,展示了细胞类型和基因功能预测的能力。在此基础上,我们认为有必要专门为非序列组学数据定制预训练工作流程,并扩展其在更广泛任务中的应用。
在这项工作中,我们通过在超过3300万个细胞上进行预训练,提出了单细胞基础模型scGPT。我们建立了一个专门用于非序列组学数据的统一生成式预训练工作流程,并调整了transformer架构以同时学习细胞和基因表示。此外,我们提供了具有特定任务目标的微调流程,旨在促进预训练模型在各种不同任务中的应用。
我们的模型scGPT通过以下三个关键方面展示了单细胞基础模型的变革性潜力:
-
scGPT代表了一个大规模的生成式基础模型,能够跨多个下游任务进行迁移学习。通过在细胞类型注释、基因扰动预测、批次校正和多组学整合等任务上达到最先进的表现,我们展示了"普适预训练,按需微调"方法作为单细胞组学计算应用的通用解决方案的有效性。 -
通过比较微调和原始预训练模型的
基因嵌入和注意力权重,scGPT揭示了关于基因-基因相互作用的宝贵生物学见解,这些相互作用与各种条件相关,如细胞类型和扰动状态。 -
我们的观察揭示了一个规模效应:更大的预训练数据量能产生更优的预训练嵌入,从而进一步提升下游任务的表现。这一发现强调了一个令人兴奋的前景:基础模型可以随着研究社群中可用测序数据的扩展而持续改进。
基于这些发现,我们预见采用预训练基础模型将大大拓展我们对细胞生物学的理解,并为未来的发现奠定坚实基础。发布scGPT模型和工作流程的目的是赋能并加速这些领域及其他领域的研究。
结果
单细胞 transformer 基础模型概述
单细胞转录组测序能够在单个细胞水平上分析分子特征。例如,scRNA-seq测量了RNA转录本的丰度,为理解细胞身份、发育阶段和功能提供了洞见。我们介绍了scGPT,一个采用生成式预训练方法的单细胞领域基础模型。核心模型包含带有多头注意力的堆叠转换器层,可以同时生成细胞和基因嵌入(方法部分)。
scGPT包含两个训练阶段:首先在大型细胞图谱上进行通用预训练,然后在较小数据集上针对特定应用进行微调(图1a-c)。在预训练阶段,我们引入了专门设计的注意力掩码和生成式训练流程,以自监督的方式训练scGPT来联合优化细胞和基因表示(方法部分)。这种技术解决了基因表达的非顺序性质,以适应NLG框架的顺序预测。在训练过程中,模型逐渐学会基于细胞状态或基因表达线索来生成细胞的基因表达。在微调阶段,预训练模型可以适应新数据集和特定任务(方法部分)。我们提供了灵活的微调流程,适用于各种基本任务,包括带批次校正的scRNA-seq整合、细胞类型注释、多组学整合、扰动预测和基因调控网络(GRN)推断。
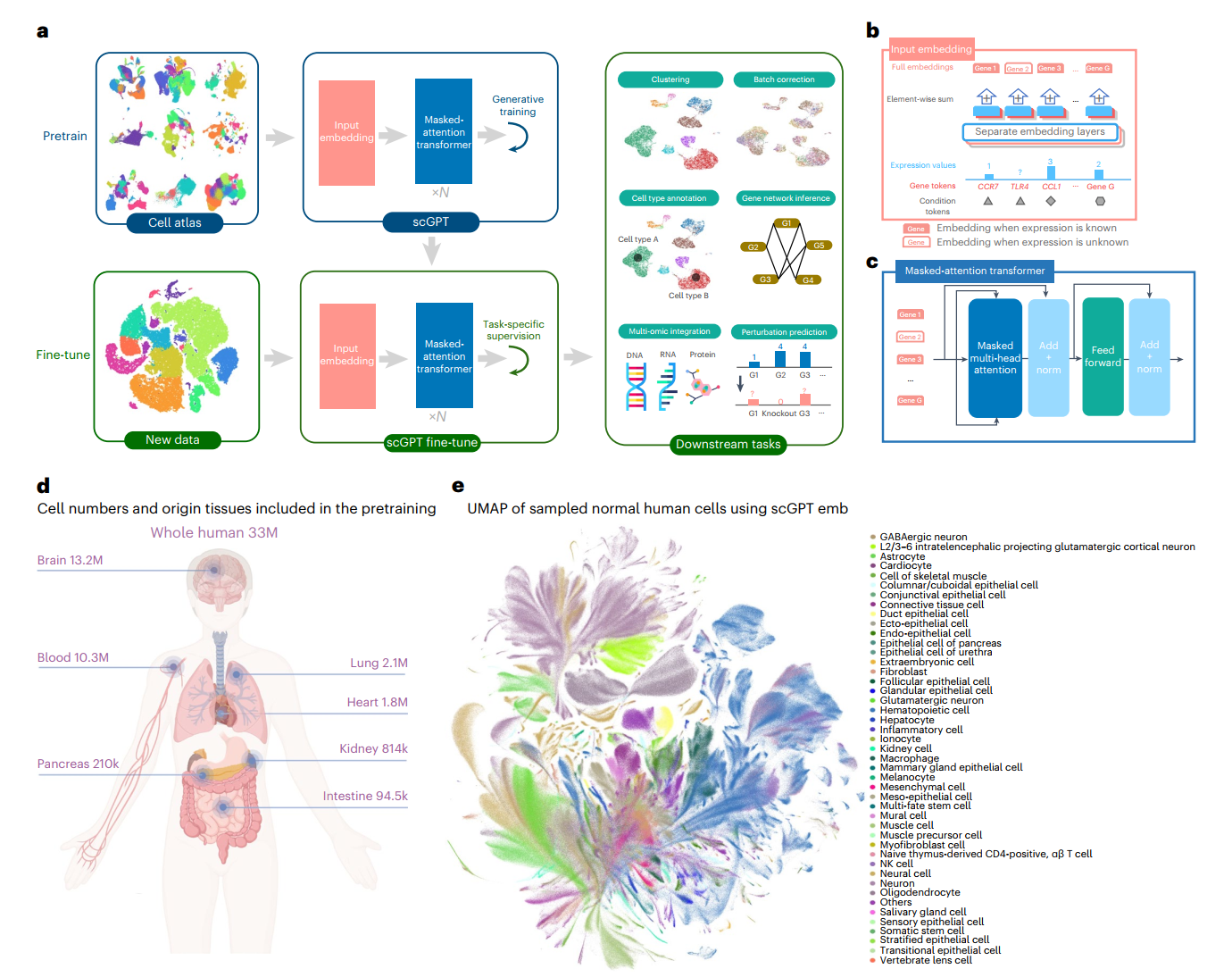
为了收集多样化和大量的测序数据用于scGPT的自监督预训练,我们从CELLxGENE收集库(https://cellxgene.cziscience.com/)组装了来自正常(非疾病)条件下3300万个人类细胞的scRNA-seq数据(图1d)。这个全面的数据集包含了来自51个器官或组织和441项研究的各种细胞类型,提供了人体中细胞异质性的丰富表示。
预训练后,我们使用统一流形近似和投影(UMAP)可视化对3300万个细胞中10%的人类细胞的scGPT细胞嵌入(图1e)。产生的UMAP图展示出引人注目的清晰度,不同颜色代表的细胞类型准确地聚集在局部区域和簇中。考虑到数据集包含超过400项研究,这表明预训练具有提取生物变异的显著能力。
UMAP (Uniform Manifold Approximation and Projection):一种非线性降维算法,通过构建高维数据的拓扑结构并将其映射到低维空间,能更好地保持数据的局部和全局结构。相比t-SNE运行更快,更适合处理大规模数据,在单细胞数据分析中应用广泛。
scGPT提高细胞类型注释的精确度
为了针对细胞类型注释微调预训练的scGPT,一个神经网络分类器以scGPT转换器输出的细胞嵌入作为输入,并输出细胞类型的分类预测。整个模型在具有专家注释的参考数据集上使用交叉熵进行训练,然后用于预测保留的查询数据分区中的细胞类型。
我们在多个数据集上进行了广泛的实验来评估scGPT在细胞类型注释方面的表现。首先,我们调整scGPT来预测人类胰腺数据集中的细胞类型。我们在图2a中可视化了预测结果。值得注意的是,scGPT对混淆矩阵中显示的大多数细胞类型都达到了较高的精确度(>0.8)(图2b),只有在参考分区中细胞数量极少的罕见细胞类型除外。例如,在10,600个参考集细胞中,肥大细胞和主要组织相容性(MHC)II类细胞类型的细胞数量少于50个。图2c可视化了微调后scGPT中的细胞嵌入,这些嵌入展示了高度的细胞类型内部相似性。
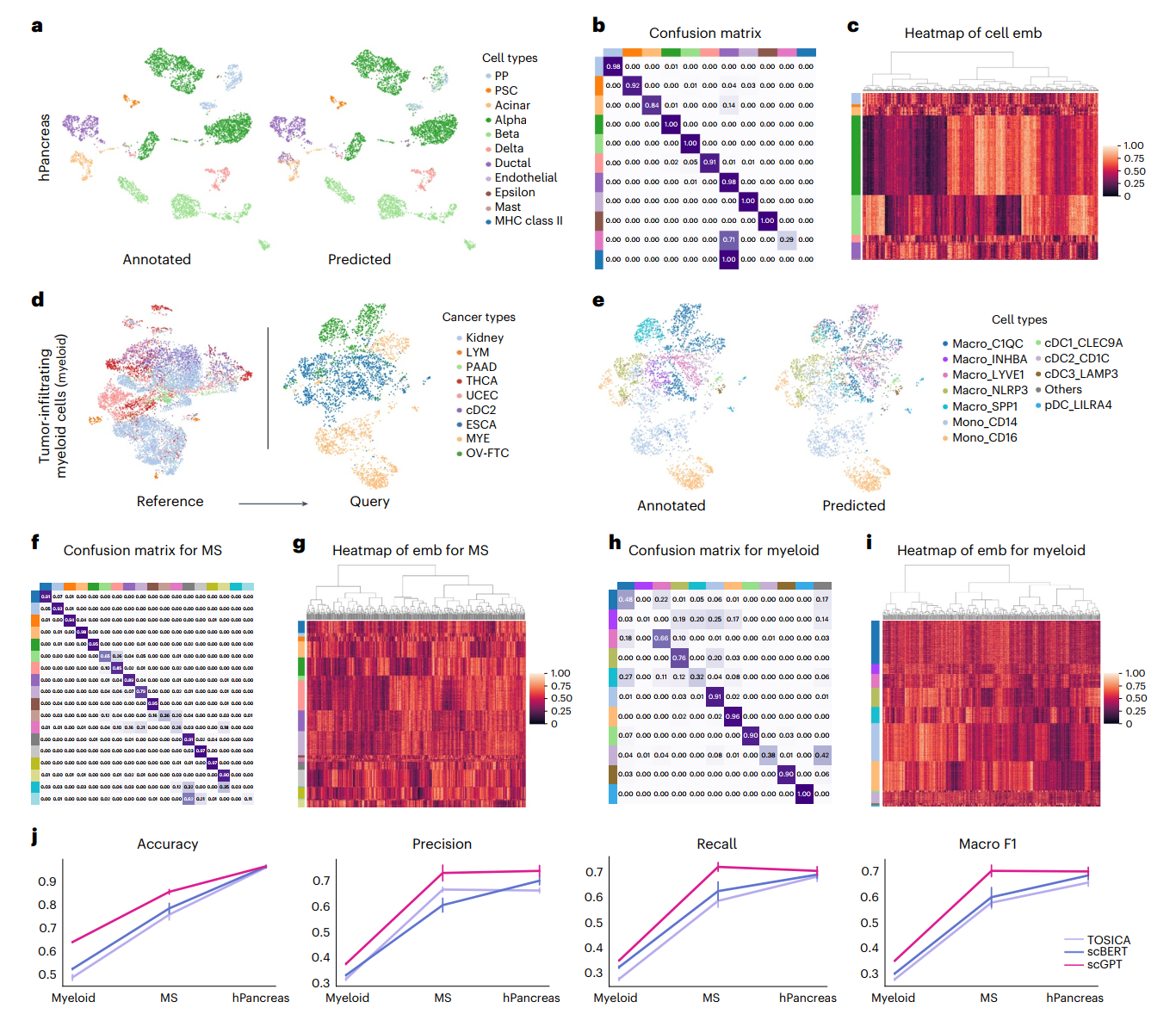
接下来,我们在一个多发性硬化症(MS)疾病数据集上测试了该模型。该模型在健康人类免疫细胞的参考分区上进行微调,并评估了对MS状态细胞的预测。微调后的模型与原始研究提供的细胞类型注释显示出强烈的一致性,并达到了约0.85的高准确度(图2f,g)。
此外,我们将该模型应用于一个更具挑战性的情况,即使用肿瘤浸润骨髓样细胞数据集来进行跨疾病类型的泛化。该模型在参考数据分区的6种癌症类型上进行微调(方法部分),并在3种未见过的癌症类型的查询分区上进行评估(图2d)。结果显示在区分免疫细胞亚型方面具有高精确度(图2e,h),并且细胞嵌入在不同细胞类型之间表现出清晰的可分性(图2i)。最后,我们在这三个数据集上对微调的scGPT与其他两种最近的基于转换器的方法TOSICA和scBERT进行了基准测试(方法部分)。scGPT在所有分类指标中,包括准确度、精确度、召回率和宏观F1值,都持续优于其他方法(图2j)。
除了细胞类型分类外,我们进一步探索了scGPT通过参考映射将未见过的查询细胞投影到参考数据集的能力(补充说明1和补充图11)。我们发现,仅使用预训练权重的scGPT就达到了与现有方法相当的性能。通过在参考数据集上进行微调可以进一步提高性能。
scGPT预测未见过的基因扰动响应
近期测序和基因编辑技术的进展极大地促进了大规模扰动实验的开展,使得我们能够表征细胞对各种基因扰动的响应。这种方法在发现新的基因相互作用和推进再生医学方面具有巨大的潜力。然而,潜在基因扰动的巨大组合空间很快就会超出实验可行性的实际限制。
为了克服这一限制,scGPT可以利用已知实验中获得的细胞响应知识,并将其外推以预测未知响应。在基因维度上使用自注意力机制,使得模型能够编码被扰动基因与其他基因响应之间的复杂相互作用。通过利用这种能力,scGPT可以有效地从现有实验数据中学习,并准确预测基因表达响应中未见过的扰动。
未见过的基因扰动预测
对于扰动预测任务,我们使用三个白血病细胞系的Perturb-seq数据集来评估我们的模型:Adamson数据集包含87个单基因扰动,经过筛选的Replogle数据集包含1,823个单基因扰动,以及Norman数据集包含131个双基因扰动和105个单基因扰动。为了评估scGPT的扰动预测能力,我们在一部分扰动上微调模型,以根据输入的对照细胞状态和干预基因来预测扰动后的表达谱。
接下来,模型在涉及未见过基因的扰动上进行测试(方法部分)。我们计算了Pearson delta指标,该指标衡量预测和观察到的扰动后表达变化之间的相关性。此外,我们还报告了每个扰动中前20个变化最显著的基因的这一指标,表示为差异表达基因上的Pearson delta。有关指标计算的详细信息,请参见补充说明12。我们对scGPT和其他两种方法GEARS和线性回归基准进行了性能比较(方法部分)。我们的结果表明,scGPT在所有三个数据集上都达到了最高分数(图3a和补充表6)。特别是,scGPT在预测扰动后的变化方面表现出色,持续超过其他方法5-20%的优势。此外,我们在图3b中可视化了Adamson数据集中两个示例扰动的预测,其中scGPT准确预测了所有前20个差异表达基因的表达变化趋势。
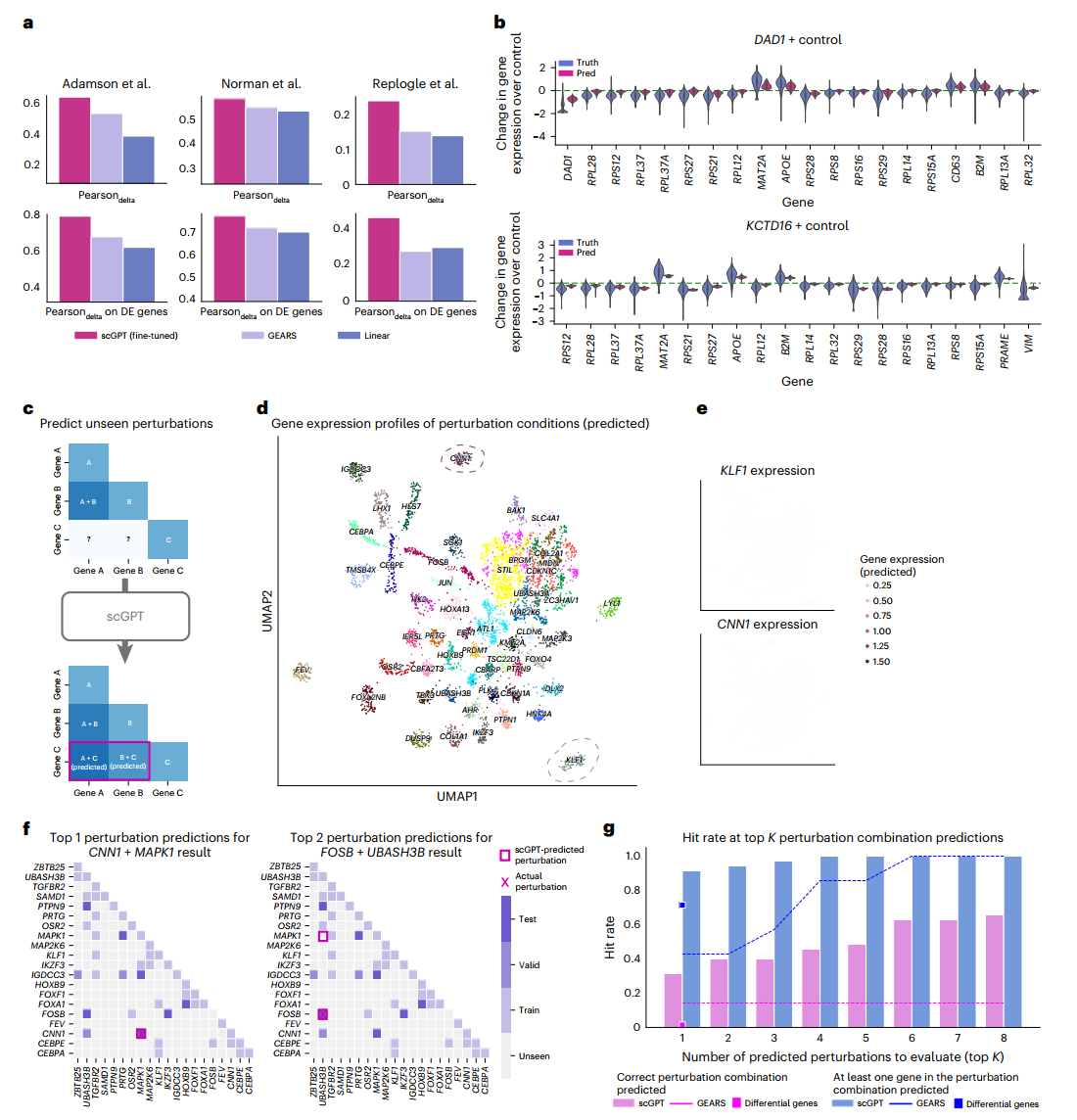
预测未见过的扰动响应的能力可以扩展扰动实验的范围,如图3c所示。为了探索预测的扰动响应的扩展空间,我们使用Norman数据集进行聚类分析,以验证生物相关的功能信号。原始的Perturb-seq研究涵盖了针对105个基因的236个扰动。
然而,考虑到这些目标基因的所有可能组合,总共有5,565个潜在的扰动,这表明实验的Perturb-seq数据仅代表了整个扰动空间的5%。因此,我们使用经过微调的scGPT来扩展扰动体外预测,并在图3d中使用UMAP可视化了每个扰动的预测平均响应。使用原始研究中的注释,我们发现相同功能组的扰动条件聚集在相邻区域(补充图4)。
接下来,我们使用Leiden对预测的表达进行聚类,并观察到这些簇与扰动组合中的"主导基因"具有高度关联。例如,与KLF1基因相关的圈出的簇表明,该簇中的数据点经历了涉及KLF1与另一个基因的组合扰动(即KLF1 + X)。以KLF1和CNN1簇为两个例子,我们进一步验证了相应的预测表达仅在这些区域高度表达(图3e),这与Norman数据集中CRISPRa(CRISPR介导的转录激活)Perturb-seq实验的预期结果一致。主导基因簇展示了scGPT揭示扰动组合之间关联的能力。
Leiden聚类:一种社区检测/聚类算法,是Louvain算法的改进版本,通常用于单细胞数据分析。它通过优化模块度(modularity)来识别网络中的社区结构,比Louvain算法更稳定,能够避免产生不连通的聚类,在保证聚类质量的同时提高运算效率。
体外逆向扰动预测
scGPT还能够根据给定的导致的细胞状态预测基因扰动的来源,我们将其称为体外逆向扰动预测。一个理想的进行这种逆向预测的预测模型可以用于推断谱系发育的重要驱动基因,或者帮助发现潜在的治疗基因靶点。
为了展示逆向扰动预测的有效性,我们使用了Norman数据集的一个子集,重点关注涉及20个基因的扰动(图3f)。这个组合空间总共包含210个单基因或双基因扰动组合。我们使用39个(18%)已知扰动(图3f中的训练组)来微调scGPT。然后,我们在未见过的扰动细胞状态的查询上测试模型,scGPT成功预测了(在排名靠前的预测中)能够产生观察结果的扰动来源。例如,scGPT将CNN1 + MAPK1基因的正确扰动排在一个测试示例的首位预测,另一个示例中将FOSB + UBASH3B基因的正确扰动排在第二位预测(图3f)。
总体而言,scGPT在前1预测中识别了平均91.4%的相关扰动(7个中的6.4个)(图3g中的蓝色条),在前8预测中识别了65.7%的正确扰动(7个测试案例中的4.6个)(图3g中的粉色条),大幅优于GEARS和差异基因基准。我们设想这些预测可以用于通过最大化达到目标细胞状态的可能性来规划扰动实验。与随机尝试相比,在这个子集中210个可能的扰动中平均需要105.5次尝试,以较少的尝试次数找到正确的基因变化来源为加速发现重要基因驱动因子和优化扰动实验提供了有价值的工具。
scGPT实现多批次和多组学整合
多批次scRNA-seq整合
整合来自不同批次的多个scRNA-seq数据集时,在保留整合数据的生物学变异的同时去除技术批次效应,这是一个独特的挑战。为了整合测序样本,我们采用自监督的方式对scGPT进行精调,通过学习统一的细胞表征来恢复被掩码的基因表达(参见方法)。
批次效应(Batch Effect):由非生物学因素造成的系统性技术偏差,例如不同实验时间、不同操作人员、不同实验室或不同仪器设备等导致的数据变异。这种效应会影响数据分析的准确性,需要通过批次校正方法来消除或减少其影响。
在我们的基准测试中,我们将scGPT与三种流行的整合方法进行比较:scVI、Seurat和Harmony。评估在三个整合数据集上进行,分别是COVID-19(18个批次)、PBMC 10k(2个批次)和大脑前嗜皮质(perirhinal cortex,2个批次)数据集。在PBMC 10k数据集中,scGPT成功区分了所有细胞类型(图4a)。scGPT出色的整合性能进一步得到了其较高生物学保留得分的支持,AvgBIO得分为0.821,比其他方法高出5-10%。AvgBIO得分汇总了三个细胞类型聚类指标:归一化互信息(NMIcell)、调整兰德指数(ARIcell)和平均轮廓宽度(ASWcell),详见补充说明12。
指标解释:
归一化互信息(NMIcell):
- 衡量两个聚类结果之间的相似度
- 值域在0-1之间,1表示完全一致
- 基于信息论,计算两种聚类方案之间共享的信息量
调整兰德指数(ARIcell):
- 评估两个聚类结果的一致性
- 考虑随机聚类的影响,对原始兰德指数进行校正
- 值域在-1到1之间,1表示完全一致,0表示随机聚类水平
平均轮廓宽度(ASWcell):
- 评估聚类的紧密度和分离度
- 计算每个样本与同类样本的相似度,与其他类样本的差异度
- 值域在-1到1之间,越接近1表示聚类效果越好
值得注意的是,即使没有精调,scGPT在整合PBMC 10k数据集时也表现出相当好的性能(补充图5),这突出了预训练的泛化能力。在近嗅皮层数据集(perirhinal cortex dataset)中,scGPT与所有其他方法相比保持竞争力(补充图6c)。这一发现突出表明,从全人类数据集学习到的特征可以很好地迁移到特定器官或组织如大脑。此外,scGPT在所有整合指标上始终保持竞争力的得分,并展现出对生物学信号的强大保留能力(补充表3和补充图6、7)。另外,我们还开发了策略来加速整合任务的精调过程,包括冻结特定模型层和排除无表达的基因,同时保持与原始方法相当的结果(补充说明3)。
单细胞多组学整合
单细胞多组学(scMultiomic)数据结合了遗传调控的多个视角,如表观遗传、转录组和翻译活性,在聚合细胞表征的同时保留生物学信号方面提出了独特的挑战。scGPT通过有效提取跨不同组学数据集的整合细胞嵌入来解决这一挑战。在10x Multiome PBMC数据集(包括联合基因表达和染色质可及性测量)的案例中,我们将scGPT与两种最新的方法进行比较,即scGLUE和Seurat(v.4)。如图4b所示,scGPT是唯一成功为CD8+ naive细胞生成独特聚类的方法。
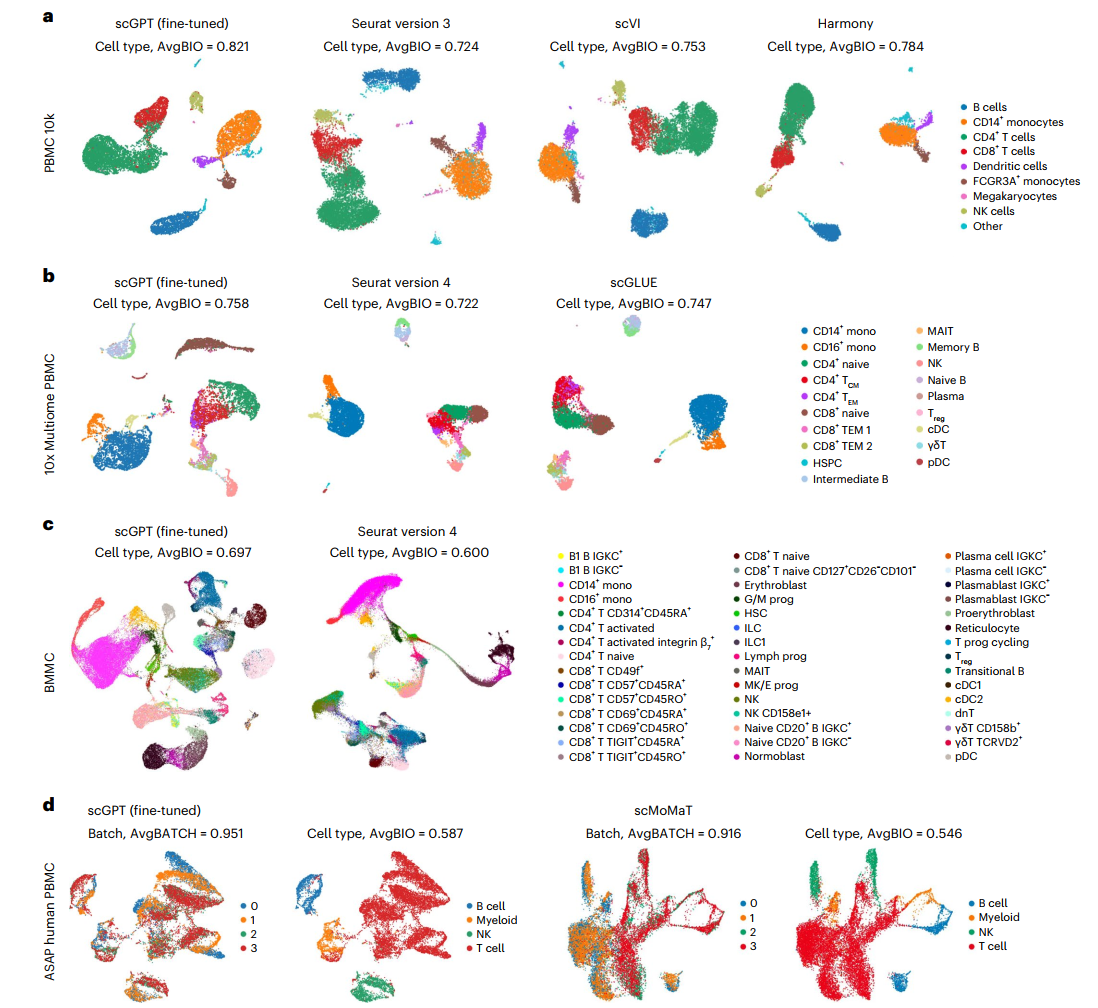
接下来,我们在来自骨髓单核细胞(BMMCs)的配对基因表达和蛋白质丰度数据集上测试了scGPT,如图4c所示。该数据集具有额外的复杂性,包括大量数据(90,000个细胞)、多个批次(12个供体)和细粒度的亚组注释(48种细胞类型)。scGPT比Seurat(v.4)呈现出更清晰的聚类结构,AvgBIO得分提高了9%。值得注意的是,scGPT能够将CD4+ naive T细胞和CD4+ activated T细胞分为两个不同的聚类。它还将整合β7+活化的CD4+ T细胞与其他CD4+ T细胞区分开来,这进一步证实了该模型能够捕捉免疫细胞亚群之间的微妙差异。
在镶嵌数据整合设置中,测序样本共享一些(但不是全部)数据模态,这给整合方法带来了挑战。为了展示scGPT在这种情况下的能力,我们以ATAC with select antigen profiling(ASAP)人类PBMC数据集为例。该数据集由四个测序批次组成,包含三种数据模态。在与scMoMat的基准实验中,scGPT展示出优越的批次校正性能,如图4d所示,特别是在B细胞、骨髓细胞和自然杀伤(NK)细胞群中。
总体而言,scGPT展示出优越的细胞类型聚类性能,并在各种基准生物学保留指标中表现出稳健性(补充表4)。
scGPT揭示特定细胞状态的基因网络
转录因子、辅因子、增强子和靶基因之间的相互作用构成了基因调控网络(GRN),调控着重要的生物学过程。现有的GRN推断方法通常依赖于静态基因表达的相关性或伪时间估计作为因果图的代理。
个人理解:研究者想知道哪个基因调控哪个基因(即因果关系),但直接观察这种调控关系很困难。所以退而求其次,主要用两种方法来推测:要么看基因表达量之间是否存在相关性(就像看到两个人总是同时出现,就推测他们可能有关系),要么通过重建细胞发育的时间顺序来推测(比如先发现A基因表达,过一会儿B基因才表达,就推测A可能调控B)。但这些都是间接的推测方法,可能并不能完全反映真实的调控关系,有点像是"猜"而不是"看到"。
scGPT通过基因表达的生成式建模进行优化,在其基因嵌入和注意力图中隐式编码了这些关系。因此,我们提出通过探测来自预训练或精调模型的scGPT嵌入和注意力图来进行GRN推断。基因嵌入构建了一个相似性网络,表明数据集层面的基因-基因相互作用。注意力图进一步捕捉了不同细胞状态下独特的基因网络激活模式。
在本研究中,我们将scGPT提取的基因网络与已知生物学进行验证,并探索其在基因程序发现中的应用。
scGPT通过学习到的基因标记嵌入,展示了其对功能相关基因进行分组和区分功能不同基因的能力。在图5a中,我们使用预训练的scGPT模型可视化了人类白细胞抗原(HLA)蛋白的相似性网络进行基本验证。在这个零样本设置中,scGPT模型成功突出了对应于两个明确特征的HLA类别的两个聚类:HLA I类和HLA II类基因。这些类别编码抗原呈递蛋白,在免疫环境中发挥不同作用。
例如,HLA I类蛋白(由HLA-A、HLA-C和HLA-E等基因编码)被CD8+ T细胞识别并介导细胞毒性效应,而HLA II类蛋白(由HLA-DRB1、HLA-DRA和HLA-DPA1编码)被CD4+ T细胞识别并触发更广泛的辅助功能。此外,我们在"免疫人类"数据集上对scGPT模型进行精调,并探索了该数据集中存在的免疫细胞类型特异的CD基因网络。我们使用与集成任务相同的精调策略(见方法)用于GRN分析。预训练的scGPT模型成功识别出了一组用于T细胞激活的T3复合物编码基因(CD3E、CD3D和CD3G)以及用于B细胞信号传导的CD79A和CD79B,以及作为HLA I类分子共受体的CD8A和CD8B(图5b)。此外,精调后的scGPT模型突出了CD36和CD14之间的联系(图5b)。
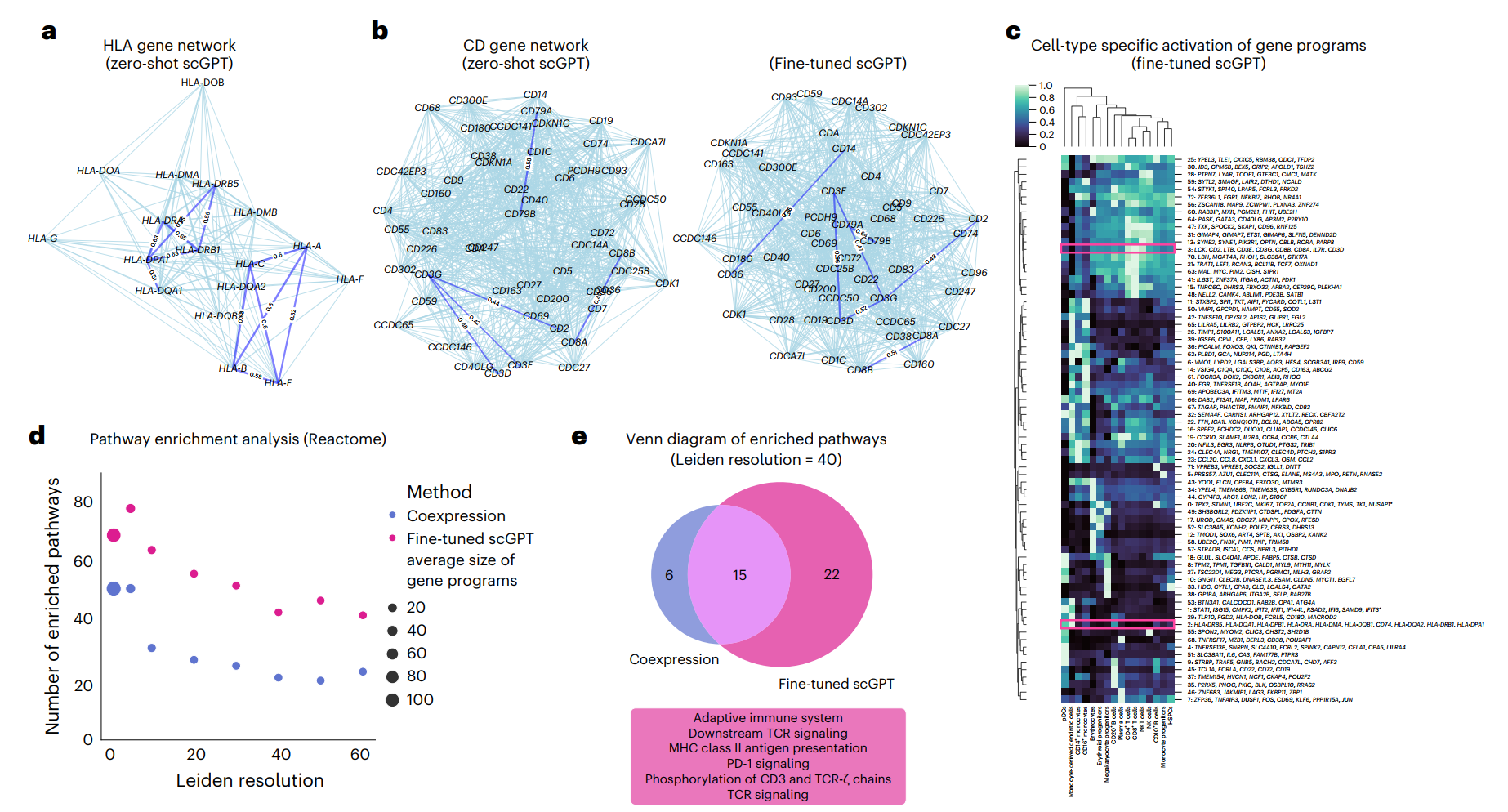
scGPT能够发现表现出细胞类型特异性激活的有意义的基因程序。基因程序随后使用来自scGPT的基因嵌入进行选择和聚类(见方法)。在图5c中,我们可视化了在"免疫人类"数据集的高变异基因(HVGs)上经过精调的scGPT模型提取的基因程序及其在不同细胞类型中的表达。我们观察到一组HLA II类基因被识别为第2组。类似地,参与T3复合物的CD3基因被识别为第3组,在T细胞中表现出最高表达。为了系统地验证提取的基因程序,我们对照Reactome数据库进行了通路富集分析,并使用严格的多重检验校正(见方法)确定了高置信度的"通路命中"。在图5d中,我们将scGPT的结果与共表达网络的结果进行比较。值得注意的是,scGPT在所有聚类分辨率下始终显示出显著更多的富集通路。
此外,我们研究了scGPT和共表达网络之间识别的通路的异同,如图5e所示。两种方法识别出15条共同通路,包括与细胞周期和免疫系统相关的通路。scGPT独特地识别出额外的22条通路,其中14条与免疫相关。值得注意的是,scGPT特别突出了与适应性免疫系统、T细胞受体信号传导、PD-1信号传导和MHC II类呈递相关的通路。这与精调数据集中存在适应性免疫群体的事实相符。这些发现证明了scGPT在捕捉复杂的基因-基因连接和揭示更广泛生物学背景下特定机制的卓越能力。富集通路的详细列表在补充表5中提供。
除了使用基因嵌入进行数据集层面的基因网络推断外,scGPT的注意力机制使其能够在单细胞层面捕捉基因-基因相互作用。scGPT通过聚合注意力图中的单细胞信号来提取细胞状态特异的网络激活数据。这为了解个别细胞内的上下文特异性基因调控相互作用提供了见解,这些相互作用可能在不同的细胞状态和条件下有所不同。
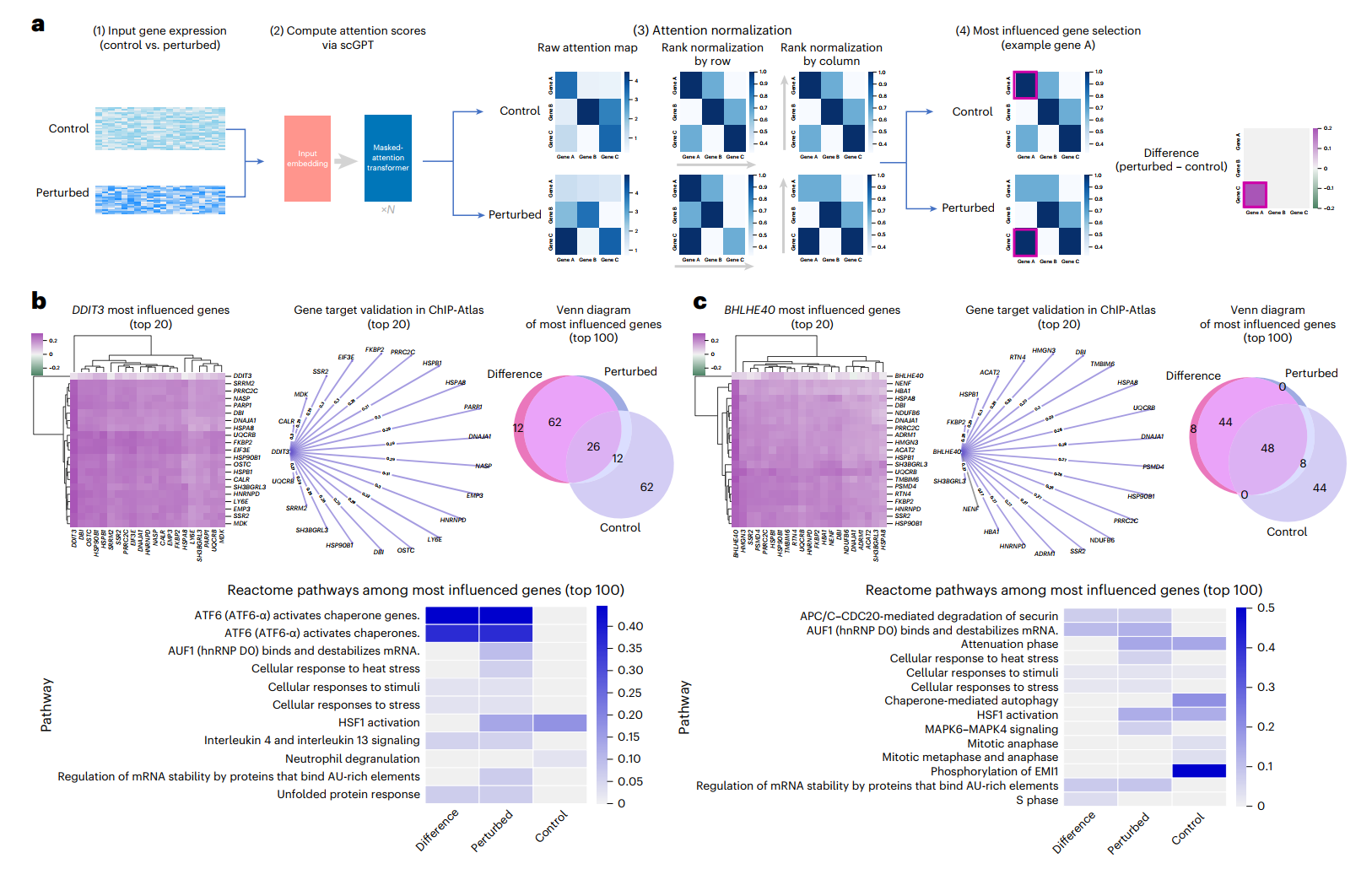
例如,在一个扰动实验中,scGPT通过检查扰动前后基因网络激活的变化,来推断哪些基因最受每个被扰动基因的影响(图6a和方法)。在Adamson CRISPR干扰数据集中,scGPT识别出了受DDIT3(编码一个转录因子)抑制影响最大的前20个基因,这些基因在ChIP-Atlas数据库中均被发现是DDIT3的信号传导靶点(图6b)。此外,scGPT在对照与DDIT3敲除设置中捕捉到了在前100个最受影响基因中不同的通路激活模式。
值得注意的是,在DDIT3敲除设置中识别出的ATF6转录因子通路已知可介导未折叠蛋白反应并调控细胞凋亡。类似地,在BHLHE40抑制的情况下,前20个最受影响的基因中有19个被发现是通过染色质免疫沉淀测序(ChIP-seq)预测的这个转录因子的靶点(图6c)。突出DNA合成和有丝分裂的通路激活模式反映了转录因子BHLHE40在细胞周期调控中的作用。这些基于注意力的发现进一步验证了scGPT在细胞状态水平上学习到的基因网络,为模型学习到的生物学提供了额外的可解释性。
迁移学习中的规模和上下文效应
在前面的章节中,scGPT通过精调的方式展示了迁移学习的巨大潜力。我们通过将其与类似的、从头训练(无预训练)的transformer模型进行比较,进一步确认了使用基础模型的好处(表示为scGPT (from scratch))。结果呈现在补充表2-4中,精调后的scGPT在整合和细胞类型注释等任务中始终表现出性能优势。鉴于基础模型对下游任务的贡献已被观察到,我们进一步关注影响迁移学习过程的因素。
首先,我们深入研究预训练数据规模与精调模型性能之间的关系:对于某个特定的分析任务,在预训练的细胞图谱中添加更多测序数据能获得多少改进?我们使用相同参数数量但使用不同数据量(从30,000到3300万个测序的正常人类细胞)预训练了一系列scGPT模型。补充图13a展示了使用这些不同预训练模型在各种应用上精调的结果表现。我们观察到,随着预训练数据量的增加,精调模型的性能有所提升(补充说明4)。这些结果表明存在一个规模效应,即更大的预训练数据规模会带来更好的预训练嵌入,并提升下游任务的性能。值得注意的是,我们的发现也与自然语言模型中报告的规模定律相一致,突出了数据规模在模型性能中的重要作用。预训练数据规模在精调结果中的关键作用表明了单细胞领域预训练模型的美好前景。随着更大、更多样化的数据集的出现,我们可以预期模型性能会进一步提升,推进我们对细胞过程的理解。
我们探索的第二个因素是上下文特定预训练的影响。这里,上下文使用是指scGPT模型在特定细胞类型上预训练,然后在类似细胞类型的下游任务上进行精调。为了探索这一因素的影响,我们在来自各个主要器官的正常人类细胞上预训练了七个器官特异性模型(图1d)和另一个泛癌症模型。我们通过可视化预训练数据的细胞嵌入来验证预训练的有效性:泛癌症模型的细胞嵌入准确地区分了不同的癌症类型(补充图2)。器官特异性模型能够揭示相应器官的细胞异质性(补充图3)。接下来,我们在COVID-19数据集上对各个模型进行微调,以检验预训练上下文的影响。我们的分析揭示了模型在预训练中所涉及的上下文相关性与其后续表现之间存在明显的关联(附录图8)。在数据整合任务中表现最好的是在全人类、血液和肺部数据集上预训练的模型,这与COVID-19数据集中存在的细胞类型高度一致。值得注意的是,尽管在1320万细胞的大规模数据集上进行了预训练,大脑预训练模型的性能仍比使用相似规模数据集训练的血液预训练模型低了8%。这强调了在预训练中保持细胞上下文与目标数据集一致的重要性,以获得更好的下游任务效果。但同时需要指出,虽然考虑细胞上下文很重要,但全人类预训练模型仍然是广泛应用场景下最可靠的选择。
讨论
我们介绍了scGPT,这是一个在大量单细胞数据上利用预训练transformer力量的基础模型。基于语言模型中自监督预训练的成功经验,我们在单细胞领域采用了类似的方法来揭示复杂的生物学相互作用。scGPT中transformer的使用使得基因和细胞嵌入的同步学习成为可能,这有助于对细胞过程的各个方面进行建模。通过利用transformer的注意力机制,scGPT能够在单细胞水平捕获基因间的相互作用,为模型提供了额外的可解释性层面。
我们通过零样本和微调环境下的全面实验展示了预训练的优势。预训练模型在零样本实验中展现出强大的推广到未见数据集的能力,呈现出与细胞类型相一致的有意义的聚类模式。此外,所学习的基因网络在scGPT中表现出与已知功能组的强烈一致性。更进一步,预训练模型的知识可以通过微调迁移到多个下游任务。在细胞类型注释、扰动预测、多批次和多组学整合等各种任务中,经过微调的scGPT模型始终优于从头开始训练的模型。这展示了预训练模型对下游任务的价值,使得分析更加准确且具有生物学意义。值得注意的是,目前的预训练并不能从本质上缓解批次效应,因此模型在具有显著技术变异的数据集上的零样本表现可能会受到限制。评估模型也很复杂,因为经常缺乏明确的生物学真值,而且数据质量也存在差异(详见附录说明10)。
对于未来的方向,我们计划在更大规模的数据集上进行预训练,包括多组学数据、空间组学和各种疾病条件。在预训练阶段纳入扰动和时序数据也很有趣,使模型能够学习因果关系,推断基因和细胞如何随时间响应变化。我们还旨在探索单细胞数据的上下文指令学习。这涉及开发使预训练模型能够在零样本环境下理解和适应不同任务和上下文的技术,而无需微调。通过使scGPT能够掌握不同分析的细微差别和具体要求,我们可以提高其在广泛研究场景中的实用性和适用性。我们设想预训练范式将很快被整合到单细胞研究中,并作为一个基础来利用来自指数增长的细胞图谱中的现有知识进行新的发现。
在线内容
任何方法、补充参考文献、《Nature Portfolio》报告摘要、源数据、扩展数据、补充信息、致谢、同行评议信息;作者贡献和竞争利益的详细信息;以及数据和代码可用性声明均可在 https://doi.org/10.1038/s41592-024-02201-0 获取。
方法
输入嵌入
单细胞测序数据被处理成一个细胞-基因矩阵,,其中每个元素 表示scRNA-seq数据的RNA分子读数或scATAC-seq数据的染色质可及性峰区域。具体来说,对于scRNA-seq数据,该元素表示细胞 中基因 的RNA丰度。在随后的章节中,我们将这个矩阵称为原始计数矩阵。scGPT的输入由三个主要组成部分:
-
基因(或峰)标记
-
表达值
-
条件标记
对于每个建模任务,基因标记和表达值都是从原始计数矩阵X相应地预处理得到的。
基因标记
在scGPT框架中,每个基因被视为类似于自然语言生成中词语的最小信息单位。因此,我们使用基因名称作为标记,并为每个基因 分配一个唯一的整数标识符 。这些标识符构成了scGPT中使用的标记词汇表。这种方法提供了极大的灵活性,可以协调具有不同基因集(即由不同测序技术或预处理流程生成)的多个研究。具体而言,不同基因标记集可以通过取所有研究中基因的并集来整合到一个共同的词汇表中。此外,我们在词汇表中还加入了特殊标记,如用于将所有基因聚合成细胞表示的<cls>和用于将输入填充到固定长度的<pad>。从概念上讲,我们将基因标记与自然语言生成中的词标记进行类比。因此,每个细胞i的输入基因标记由一个向量 表示:
其中M是预定义的最大输入长度。
表达值
在用于建模之前,基因表达矩阵X需要额外的处理。基因表达建模中的一个基本挑战是不同测序协议间绝对量值的变异性。测序深度的差异以及稀疏表达基因的存在导致了不同测序批次样本之间的数据尺度存在显著差异。这些差异不容易通过常见的预处理技术(如每百万转录本归一化和log1p转换)来缓解。即使经过这些转换,相同的绝对值在不同测序批次间也可能传达不同的"语义"含义。为了解决这种尺度差异,我们提出了值分箱技术,将所有表达计数转换为相对值。对于每个细胞中的每个非零表达计数,我们计算原始绝对值并将它们分成B个连续区间 ,其中 。每个区间代表所有表达基因的相等部分(1/B)。需要注意的是,每个细胞都会计算一组新的分箱边界,因此区间边界可能在细胞间有所不同。细胞i的分箱表达值 定义为:
通过这种分箱技术, 的语义含义在来自各种测序批次的细胞间保持一致。例如, 始终表示基因中的最高表达。值得注意的是,对于微调任务,我们还在值分箱步骤之前执行了log1p转换和高变异基因选择。为了简化表示,我们使用 来表示分箱前的原始和预处理数据矩阵。因此,细胞i的分箱表达值的最终输入向量表示为:
条件标记
条件标记包含了与各个基因相关的多样化元信息,比如扰动实验的改变(由扰动标记指示)。为了表示位置相关的条件标记,我们使用一个与输入基因具有相同维度的输入向量。该向量表示为:
其中 表示对应于某个条件的整数索引。
嵌入层
我们使用常规的嵌入层(即PyTorch嵌入层,https://pytorch.org/docs/stable/generated/torch.nn.Embedding.html) 和 分别用于基因标记和条件标记,以便将每个标记映射到维度D的固定长度嵌入向量。我们使用全连接层(表示为 )来处理分箱表达值以增强表达能力。这种选择使得对基因表达值的序数关系进行建模成为可能。因此,细胞i的最终嵌入 定义为:
细胞和基因表达建模的transformer
scGPT transformer
我们使用自注意力transformer来编码方程(5)中的完整输入嵌入 。自注意力机制作用于M个嵌入向量序列,这使其特别适合捕获基因之间的相互作用。堆叠的transformer块的输出可以定义如下:
我们使用得到的表示 来进行基因级别和细胞级别的任务。基因级别的微调目标直接应用。例如包括基因表达预测(GEP)目标和扰动表达预测任务(perturb-GEP)。对于细胞级别的任务,我们首先将 整合成一个细胞嵌入向量。一个例子是细胞类型分配任务,其中细胞嵌入用于通过在细胞类型分类训练目标中添加的分类器来预测细胞类型标签。
输入维度M可以达到数万个基因,这大大超过了自然语言生成中常用的传统transformer的输入长度。为了解决这个挑战并确保高效的自注意力机制,我们利用FlashAttention的加速自注意力实现。这种实现有效地提高了模型容量,并能够有效处理大输入维度。尽管我们采用了FlashAttention,但任何高效的transformer都可能用于scGPT,例如具有线性复杂度的transformer(Linformer)和Kernelized Self-Attention(KSA)。
细胞表示
每个细胞类似于由基因组成的"句子",其表示 是通过聚合学习到的基因级别表示 获得的。
各种池化操作,如逐元素平均池化或加权池化,都可以在这种情况下轻松使用。
在本研究中,我们选择使用特殊标记<cls>来获取细胞表示,使模型能够在transformer块内学习池化操作。<cls>标记被添加到输入标记的开头,这个位置的最终嵌入被提取作为细胞表示。因此,细胞嵌入 可以通过从堆叠的最终层嵌入 中提取相应行来获得,其中 操作检索<cls>标记位置的行。
批次和模态的表示
我们使用额外的一组标记来表示不同的测序批次和测序模态(来自RNA-seq的基因、来自ATAC-seq的峰等),特别是对于scRNA-seq和scMultiomic整合任务。这类似于输入嵌入中介绍的条件标记,并且使用标准嵌入层以类似的方式实现。模态标记 与个别输入特征 相关联(例如,指示它是基因、区域还是蛋白质)。批次标记最初是在细胞水平上的,但也可以传播到单个细胞的所有特征。换句话说,相同的批次标记 可以重复到单个细胞i的输入特征长度M:
和批次及模态表示的区别在于,这些批次和模态嵌入并不作为输入送入转换器模块。相反,它们在进入特定的微调目标之前,要么在特征层面或者细胞层面与转换器的输出进行拼接。这样做是为了防止转换器在同一模态的特征内放大注意力,同时低估不同模态之间的注意力。
此外,在下游微调目标中了解模态和/或批次标识有助于基因表达建模。当模型学会基于模态和/或批次标识预测表达值时,这些偏差会从基因和细胞表示本身中被隐式地消除。这可以作为一种促进批次校正的技术。
比如,在scMultiomic整合任务中,我们将转换器输出与批次和模态嵌入的和进行拼接。这作为下游微调目标的输入用于表达建模:
其中 和 分别表示批次和模态嵌入层, 表示转换器层的输出(scGPT transformer)。
另外,在scRNA-seq整合任务中,批次嵌入与细胞表示的拼接产生以下作为输入的表示:
其中 表示细胞i的批次标识。是原始细胞表示(微调目标)。需要注意的是,修改后的版本 仅与表达建模目标相关,不适用于基于分类的目标,详见微调目标部分。
生成式预训练
基础模型预训练
基础模型被设计成一个可泛化的特征提取器,能够服务于多种下游任务。预训练中使用的标记词汇包含人类基因组中的全部基因集。在模型预训练之前,表达值被进行了分箱处理(输入嵌入)。为了加快训练速度,我们限制每个输入细胞只包含非零表达的基因。为了有效地训练模型以捕获基因-基因关系和基因-细胞关系,我们引入了一个带有特殊注意力掩码的生成式训练策略,具体将在下一节描述。
用于生成式预训练的注意力掩码
自注意力机制已被广泛用于捕获标记之间的共现模式。在自然语言处理中,这主要通过两种方式实现:
-
掩码标记预测,用于
BERT和RoBERTa等转换器编码器模型,其中输入序列中随机掩码的标记在模型输出中被预测; -
在因果转换器解码器模型(如
OpenAI GPT系列)中使用序列预测的自回归生成。
OpenAI GPT-3和GPT-4中使用的生成式预训练采用了一个统一的框架,其中模型根据由已知输入标记组成的"提示"来预测最可能的下一个标记。这个框架在各种自然语言生成应用中提供了极大的灵活性,并在零样本和微调设置中展示了上下文感知等能力。我们相信生成式训练同样可以以类似的方式有益于单细胞模型。具体来说,我们对两个任务感兴趣:
-
基于已知的基因表达生成未知的基因表达值,即通过"
基因提示"进行生成; -
给定输入细胞类型条件生成全基因组表达,即通过"
细胞提示"进行生成。
尽管标记和提示的概念相似,但由于数据的非序列性质,对基因读数的建模本质上与自然语言不同。与句子中的单词不同,细胞内基因的顺序是可互换的,并且没有等效的"下一个基因"预测概念。这使得直接将GPT模型中的因果掩码公式应用于单细胞数据变得具有挑战性。为了解决这个挑战,我们为scGPT开发了一种特殊的注意力掩码机制,根据注意力分数定义预测顺序。
注意力掩码通常可以应用在转换器模块中的自注意力图上:对于M个基因标记的输入(公式(1)),第 个转换器模块对其输入 的M个标记应用多头自注意力(公式(6,7))。具体来说,每个自注意力操作计算如下:
其中 代表查询、键和值向量。 是可学习的权重矩阵。 是特征维度,作为 中的缩放因子以维持数值稳定性。注意力掩码 通过修改查询和键之间的原始注意力权重 来划定自注意力的范围。
transformer模型中的相关概念可参考这篇文章:
具体来说,在矩阵中位置 添加 -inf 会在 softmax 后使注意力权重归零,从而禁止第i个查询和第j个键之间的注意力。另一方面,添加0意味着注意力权重保持不变。这种注意力掩码技术允许模型专注于特定的上下文元素。
我们专门设计了scGPT注意力掩码,以统一的方式支持基因提示和细胞提示生成。注意力掩码 在附图1a中进行了可视化,其中查询按行排列,键按列排列。如图底部注释所示,输入嵌入 中的每个标记可以属于以下三组之一:
(1)用于细胞嵌入的保留<cls>标记(在"细胞表示"小节中介绍);
(2)具有标记嵌入和表达值嵌入的已知基因;
(3)需要预测表达值的未知基因。
scGPT注意力掩码的基本规则是只允许"已知基因"的嵌入和查询基因本身之间的注意力计算。这通过使用 中的元素 实现如下:
在每次生成迭代中,scGPT预测一组新基因的基因表达值,这些基因反过来在下一次迭代的注意力计算中成为"已知基因"。这种方法通过在非序列单细胞数据中进行顺序预测,反映了传统转换器解码器中带有下一个标记预测的因果掩码设计。
如附图1a所示,在训练期间,我们随机选择一定比例的基因作为未知基因,以便在输入中省略其表达值。注意力仅在已知基因和查询未知基因本身之间应用,而不应用于其他未知基因的位置。
例如,在位置j要预测的基因只与细胞嵌入、已知基因和它自身有注意力分数,而不与其他未知基因有注意力分数,如注意力掩码的最后一行所示。scGPT模型通过带有上述掩码注意力图的堆叠转换器模块来预测这些未知基因的表达。推理步骤如附图1b所示。在细胞提示生成的推理过程中,scGPT基于特定的细胞类型条件生成全基因组表达。在第一个位置输入代表细胞类型条件的训练好的细胞嵌入。数千个基因表达值的整个生成过程在K个迭代步骤中进行(例如,附图1b中的K=3步)。
例如,在第 次迭代中,注意力掩码机制允许与前面 0 到 次迭代中所有预测的基因进行注意力计算。在每次迭代中,scGPT从未知集合中选择预测置信度最高的前 个基因,作为下一次迭代 的已知基因。
直观地说,这个工作流以自回归方式简化了基因表达的生成,其中首先生成预测置信度最高的基因表达值,并用于帮助后续的生成轮次。基因提示生成以类似的迭代方式工作。不同之处在于,它从一组具有观察到的表达值的已知基因开始,而不是从细胞嵌入开始。
scGPT注意力掩码统一了已知基因的编码过程和未知基因的生成。它也是首批针对非序列数据进行自回归生成的转换器方案之一。
学习目标和预训练
我们使用了一个基因表达预测目标来优化模型,以预测未知基因的表达值。具体来说,我们使用多层感知机网络(MLP)来估计未知表达值并计算均方损失:
其中 表示未知基因的输出位置集合,是待预测的实际基因表达值。操作获取集合中元素的数量。
如"用于生成式预训练的注意力掩码"小节中所述,支持基因提示和细胞提示两种生成模式。在训练过程中,这两种模式连续进行。在一个给定细胞的输入基因标记中,选择一定比例的基因作为"未知"基因,并省略它们的表达值。首先,在基因提示步骤中,模型的输入包含<cls>标记嵌入、已知基因嵌入和未知基因嵌入。使用模型的输出计算损失(公式(14))。其次,在细胞提示步骤中,上一步的输出细胞嵌入(即"细胞表示"中的 )用于替换<cls>位置的嵌入。其他计算保持不变。最后,将这两个步骤的损失值加在一起,用于计算梯度以优化模型参数。
微调目标
scGPT利用各种微调目标来促进对细胞和基因的生物学有效表示的学习,以及用于批次校正等正则化目的。
基因表达预测
为了促进基因-基因互作的学习,scGPT包含了基因表达预测(GEP)。这个微调目标的工作方式与预训练中的目标("学习目标和预训练")类似,但应用于掩码位置。具体来说,对于每个输入细胞,随机掩码一部分基因标记及其对应的表达值 。scGPT被优化以准确预测掩码位置的表达值。这个微调目标有助于模型有效编码数据集中基因之间的共表达。该目标最小化掩码位置(表示为)的均方误差。GEP的工作方式如下:
这里,表示细胞i的表达估计行。值得注意的是,如果提供了测序批次或模态条件,我们使用公式(9)中的 而不是 。
GEP提供了一个通用的自监督微调目标,旨在预测基因表达值。在某些下游任务中,例如扰动预测,模型需要预测扰动后的基因表达值而不是原始值。我们将这种变体称为perturb-GEP。我们保留公式(15)和公式(16)中的MLP估计器,但使用扰动后的基因表达作为目标 。在perturb-GEP中,模型应该预测所有输入基因的扰动后表达。
用于细胞建模的基因表达预测
这个微调目标的操作方式与GEP类似,但基于细胞表示 预测基因表达值,以明确促进细胞表示学习。对于输入细胞i中的每个基因j,我们创建一个查询向量 ,并使用 和细胞表示 的参数化内积作为预测的表达值:
用于细胞建模的GEP(GEPC)继承了公式(5)中的基因标记嵌入 。在整合任务中,我们使用公式(10)中的 而不是 。在我们的实验中,我们观察到将GEP和GEPC结合使用与单独使用任一方法相比,性能有显著提升。
弹性细胞相似度
这个微调目标通过使用相似度学习损失来增强细胞表示:
其中 表示余弦相似度函数,而 和 指的是小批量内的两个细胞。此外, 表示预定义的阈值, 是弹性细胞相似度。这种方法的基本思想是增强相似度值高于 的配对之间的相似性,从而使它们更加相似。相反,不相似的配对则被鼓励进一步分开。
通过反向反向传播进行域适应
由于测序技术引入的非生物学批次差异导致的批次效应,会妨碍细胞表示的学习。为了缓解这个问题,我们使用一个独特的MLP分类器来预测每个输入细胞的测序批次(基于它们的细胞表示 ),并通过在模型内反转梯度来修改反向传播过程。这种方法借鉴了Ganin和Lempitsky提出的鲁棒域适应方法的见解。
细胞类型分类
这个微调目标旨在利用学习到的细胞表示来注释单细胞。我们使用一个单独的MLP分类器从细胞表示 中预测细胞类型。这个微调目标通过预测的细胞类型概率和真实标签之间的交叉熵损失ce进行优化。
下游任务的微调
细胞类型注释
对于细胞类型注释任务,我们在具有真实标签的参考集上对模型进行微调,并在保留的查询集上验证注释性能。保留预训练基础模型和参考集之间的共同基因标记集。在模型微调之前,基因表达值经过归一化、对数转换和分箱处理。除了输出细胞类型分类器(随机初始化)外,所有预训练模型权重都用于初始化微调模型。在训练中使用所有具有零和非零表达值的基因标记。使用细胞类型分类微调目标来最小化分类损失。
基因扰动响应预测
为了针对扰动预测任务进行微调,我们在模型训练前选择了高变异基因并对表达值进行了预处理。嵌入层和transformer层的参数从预训练模型中初始化。在微调过程中,包含了所有零值和非零值表达的基因标记。针对扰动预测任务的输入进行了两个显著的改变:
首先,我们使用了log1p转换后的表达值作为输入和目标值,而不是分箱值,以更好地预测扰动后的绝对表达;其次,我们在每个输入基因位置添加了二元条件标记,以指示该基因是否被扰动。
我们采用了perturb-GEP微调目标,并对训练设置进行了进一步修改。不同于使用同一细胞的遮罩和未遮罩表达值作为输入和学习目标,我们使用对照细胞作为输入,扰动细胞作为目标。这是通过将每个扰动细胞与随机配对的非扰动对照细胞构建输入-目标对来实现的。输入值包含了对照细胞中所有基因的表达值。因此,模型学会了基于对照基因表达和扰动标记来预测扰动后的反应。
整合多个scRNA-seq数据集的批次校正
当输入原始计数矩阵包含来自不同测序批次或技术的多个数据集时,批次效应可能是细胞类型聚类中的一个主要混淆因素。因此,在整合多个scRNA-seq数据集时,我们的目标是在保持生物学差异的同时校正批次效应。对于这个整合任务的微调,我们保留了预训练基础模型和当前数据集之间的共同基因标记集。我们进一步从共同集中选择了一部分HVGs作为输入。我们在模型训练前对表达值进行了预处理,类似于细胞类型注释任务。所有预训练模型权重都被用来初始化微调模型。默认情况下,训练中使用了所有具有零值和非零表达值的基因标记。除了GEP和GEPC之外,ECS、通过反向传播的域适应(DAR)和DSBN微调目标被同时优化,通过反向传播和特定域归一化来增强细胞对比学习和显式批次校正。
scMultiomic数据的整合表示学习
scMultiomic数据可能在实验批次间包含不同的测序模态。我们研究了scMultiomic数据的两种数据整合设置:配对设置(paired)和镶嵌设置(mosaic)。
在配对设置中,所有样本(细胞)共享所有测序的数据模态。在镶嵌设置中,一些批次共享少数共同的数据模态,但不是全部。由于存在额外的ATAC(和/或)蛋白质标记,我们仅继承了RNA数据的训练基因嵌入,并从头开始训练额外的标记嵌入和模型的其余部分。如果数据集包含额外的蛋白质数据,训练中仅使用非零表达值的标记。否则,默认情况下使用零值和非零表达值。我们使用了一组额外的模态标记来指示每个标记的数据类型(即基因、区域或蛋白质),并促进GEP和GEPC微调目标中的掩蔽基因和值预测。默认情况下,模型使用GEP和GEPC微调目标进行优化。如果存在多个批次,则包含DAR以促进多模态批次校正。
基因调控网络推断
对于图5中基于基因嵌入的GRN推断,在零样本设置中,我们基于k-最近邻从scGPT的预训练基因嵌入构建了基因相似性网络。在微调设置中,我们以类似的方式从在人类免疫数据集上微调的scGPT模型构建了基因相似性网络。我们进一步对相似性图进行了Leiden聚类,并从包含五个或更多基因的基因簇中提取基因程序。
对于图6中基于注意力的目标基因选择,我们在Adamson扰动数据集上微调了scGPT血液模型,该数据集包含了在白血病细胞系上进行的87个CRISPR干扰实验。我们在图6a中展示了目标基因选择流程。对于每个感兴趣的扰动基因,我们首先通过分别输入扰动和对照细胞集,获得了两组注意力图谱(扰动组和对照组)。
注意,原始注意力分数是从模型最后一个注意力层的所有8个注意力头中获得的。原始注意力分数随后经过两轮秩归一化,先按行后按列进行。然后对8个注意力头的秩归一化注意力分数进行平均,得到一个聚合注意力图谱。这就得到了用于最受影响基因选择的最终注意力图谱。对于每个感兴趣的扰动基因,我们通过对扰动基因所在列中的最终注意力图谱中的分数进行排序来选择其最受影响的基因。这反映了注意力图谱中的列表示感兴趣的基因对其他基因的影响程度这一直觉。我们提供了三种最受影响基因选择设置:来自对照注意力图谱的"对照"、来自扰动注意力图谱的"扰动"以及两者之差的"差异"。从对照注意力图谱选择的基因靶标应反映感兴趣基因参与的基础通路,而扰动注意力图谱则反映扰动后的效应。这两个注意力图谱之间的差异应突出显示从扰动前到扰动后基因网络中发生最大变化的边。
同样地,对于涉及多个转录因子的扩展注意力基因相互作用预测(补充说明7),我们在Replogle数据子集上微调了scGPT血液模型,并报告了来自"扰动"设置的最受影响基因。
数据集
CELLxGENE scRNA-seq集合
我们从CELLxGENE门户网站(https://cellxgene.cziscience.com/)使用Census API收集了用于整个人类基础模型预训练的数据(Census API可在 https://chanzuckerberg.github.io/cellxgene-census/python-api.html 获取,它定期托管和更新在线数据发布。我们使用了2023年5月15日的发布版本)。我们包含了scRNA-seq和snRNA-seq的测序协议,并过滤出没有疾病状况的样本。这最终得到了3300万个细胞的测序数据。
特别地,为了预训练scGPT血液模型,我们从CELLxGENE(https://cellxgene.cziscience.com/)获取了超过1030万个人类血液和骨髓scRNA-seq样本。通过对生物体(即Homo sapiens)、组织(即血液、骨髓)和疾病(即正常、COVID-19、流感)进行过滤,从CELLxGENE中总共收集了65个数据集。此外,我们收集了570万个各种癌症类型的细胞来训练泛癌症模型。
多发性硬化症
MS数据集从EMBL-EBI(https://www.ebi.ac.uk/gxa/sc/experiments/E-HCAD-35)获取。数据集包含了9个健康对照样本和12个MS样本。我们将对照样本划分用于模型微调的参考集,并将MS样本作为查询集用于评估。这个设置作为分布外数据的示例。我们排除了三种细胞类型:B细胞、T细胞和少突胶质前体细胞,这些细胞类型仅存在于查询数据集中。最终的细胞数量在训练参考集中为7,844个,在查询集中为13,468个。使用原始发表文献中提供的细胞类型标签作为评估的真实标签。数据处理协议涉及选择HVGs以保留3,000个基因。
骨髓样细胞
骨髓样数据集可从基因表达综合数据库(GEO)使用登录号 GSE154763 获取。该数据集包含九种不同的癌症类型,但为了训练和评估模型,六种癌症类型被选作参考集用于训练,而三种癌症类型用作查询集。参考集包含骨髓癌症类型UCEC、PAAD、THCA、LYM、cDC2和kidney,而查询集包含MYE、OV-FTC和ESCA。数据集也进行了随机抽样。最终的细胞数量在参考集中为9,748个,在查询集中为3,430个。在数据处理过程中选择了3,000个HVGs。
人胰腺
人胰腺数据集包含来自五个人胰腺细胞scRNA-seq研究的数据,这些数据由Chen等人重新处理用于细胞类型注释任务。这五个数据集按数据来源分为参考集和查询集。参考集包含来自两个数据源的数据,查询集包含其他三个数据源的数据。参考集和查询集都保留了3,000个基因和来自原始发表文献的真实注释。参考集包含13个细胞群的10,600个细胞(alpha、beta、导管、腺泡、delta、胰腺星状、胰腺多肽、内皮、巨噬细胞、肥大细胞、epsilon、施旺细胞和T细胞)。查询集包含11个细胞群的4,218个细胞(alpha、beta、导管、胰腺多肽、腺泡、delta、胰腺星状、内皮、epsilon、肥大细胞和MHC II类)。
PBMC 10k
PBMC 10k数据集包括来自一位健康供体的两批scRNA-seq人类外周血单个核细胞。该数据集由Gayoso等人重新处理,得到3,346个差异表达基因。第一批包含7,982个细胞,第二批包含4,008个细胞。使用Seurat注释的细胞群包括九个类别,即B细胞、CD4+ T细胞、CD8+ T细胞、CD14+单核细胞、树突状细胞、NK细胞、FCGR3A+单核细胞、巨核细胞和其他。
人类免疫
人类免疫数据集包括五个scRNA-seq数据集:一个来自人类骨髓,四个来自人类外周血。使用了各种测序技术,包括10x Genomics、10x Genomics (v.2)、10x Genomics (v.3)和Smart-seq2。该数据集总共包含33,506个细胞和12,303个基因。根据供体来源定义了十个不同的批次。协调后的数据包含16个细胞群。我们使用了由Luecken等人重新处理的数据和注释。
近嗅皮层(Perirhinal cortex)
近嗅皮层数据集包括两个不同的样本,取自Siletti等人的一项更大规模研究,该研究最初包含606个高质量样本,涵盖十个不同的脑区。从近嗅皮层数据集选取的两个批次都包含大量细胞,第一批包含8,465个细胞,第二批包含9,070个细胞。这些数据集包含了范围广泛的59,357个基因。我们使用了原始研究中提供的十种独特细胞类型的注释。
COVID-19
COVID-19数据集来源于Lotfollahi等人的工作,分为18个不同的批次,包含来自肺组织、PBMCs和骨髓的多样化细胞。该数据集最初包含274,346个细胞和18,474个基因,为了本研究的目的,已被抽样至总共20,000个细胞。我们使用了原始研究提供的注释。对于参考映射评估,我们随机选择了12个样本批次作为参考数据集,另外6个批次作为查询数据集。最终的参考数据集包含15,997个细胞,查询数据集包含4,003个细胞。
Adamson
Adamson扰动数据集包含通过Perturb-seq扰动的K562白血病细胞系的基因表达数据。该数据集包括87个独特的单基因CRISPR干扰扰动,每个扰动在大约100个细胞中重复。
Norman
Norman扰动数据集包含通过Perturb-seq扰动的K562白血病细胞系的基因表达数据。该数据集有131个双基因扰动和105个单基因扰动。每个扰动在约300-700个细胞中重复。
Replogle
Replogle扰动数据集包含对K562白血病细胞系进行CRISPR干扰的全基因组扰动。考虑到数据质量,我们保留了与原始研究中识别的1,973个具有强转录表型扰动相匹配的数据子集。我们还删除了150个在测序数据中没有扰动基因表达记录的扰动。我们进一步为每个扰动保留了100个样本和2,500个对照样本。处理后的完整数据集包括来自1,823个扰动的171,542个样本,其中99个是转录因子的扰动。测试集包括456个扰动,其中25个是转录因子的扰动。
Multiome PBMC
10x Multiome PBMC 数据集包含来自人类PBMC细胞的配对单细胞RNA和ATAC数据,这些数据是通过10x Single Cell Multiome协议测序获得的。在该数据集中,所有样本都来自同一位健康供体。每个细胞都具有基因表达和染色质可及性测量。处理后的数据来自9,631个细胞,包含来自29,095个基因和107,194个染色质区域的读数计数。注释包括19个细胞群(CD14+单核细胞、CD16+单核细胞、CD4+初始、CD4+ TCM、CD4+ TEM、CD8+初始、CD8+ TEM 1、CD8+ TEM 2、HSPCs、中间B细胞、MAIT、记忆B细胞、NK、初始B细胞、浆细胞、Treg、cDC、gdT和pDC)。
BMMC
BMMC数据集包含通过CITE-seq协议测序的BMMCs配对单细胞RNA和蛋白质丰度测量。这些细胞来自12位健康人类供体,在该数据集中构成12个批次。处理后的数据代表90,261个细胞,包含来自13,953个基因和134个表面蛋白的测量。注释包含45个详细的免疫细胞亚型。
ASAP PBMC
ASAP PBMC数据集包含四个测序批次,具有三种数据模态(基因表达、染色质可及性和蛋白质丰度)。四个批次分别包含5,023、3,666、3,517和4,849个细胞。在批次1和2中,所有样本都有来自CITE-seq的4,768个基因和216个蛋白质测量。在批次3和4中,所有样本都有17,742个区域和来自ASAP-seq的相同216个蛋白质测量。注释包含四个细胞群(B细胞、髓样细胞、NK细胞和T细胞)。
Lung-Kim
该数据集包括14个原发性人类肺腺癌样本,总共32,493个细胞。该数据集可通过策展癌症细胞图谱公开访问。对于我们的研究,我们将其分为十个样本的参考集和四个样本的查询集。最初,该数据集包括11个细胞群:B细胞、树突状细胞、内皮细胞、上皮细胞、成纤维细胞、巨噬细胞、恶性细胞、肥大细胞、NK细胞、T细胞和一个未确定类别。在预处理中,我们移除了未确定类别并选择了3,000个HVGs用于下游评估。最终数据集包含30,472个细胞。对于参考映射评估,我们随机选择了十个患者样本作为参考数据集,另外四个患者样本作为查询数据集。最终的参考数据集包含24,746个细胞,查询数据集包含7,747个细胞。
基准测试实验设置
单细胞RNA测序细胞类型注释
我们在骨髓、多发性硬化症和人类胰腺数据集上,将scGPT与两种最新的基于transformer的细胞类型注释方法(scBERT和TOSICA)进行了基准测试对比。对于每个数据集,如前文所述,我们使用参考数据划分进行模型训练和验证。对查询集预测的细胞类型标签用于评估。
我们基于四个标准分类指标评估细胞类型分配的性能:准确率、精确率、召回率和宏观F1值。准确率、精确率和召回率是针对整体性能进行全局计算,而宏观F1值则是按类别平均以增加稀有细胞类型的权重。我们还报告了按细胞类型"精确率"的归一化混淆矩阵以提供更多细节。
单细胞RNA测序扰动
我们将scGPT与最新的扰动预测方法GEARS和线性回归模型进行了比较。线性回归模型将对照细胞的基因表达值和每个基因的扰动状态二进制编码作为输入特征。该模型通过最小化回归误差,使用输入特征的线性组合来估计每个基因的扰动后表达。为确保一致性,我们遵循了Roohani等人在其基准测试中概述的预处理步骤。将25%的扰动划分为测试集,在训练期间保持未见。
虽然GEARS研究报告CPA方法表现次优,我们在实验中证实了这些发现。然而,我们认为这种性能差距主要归因于实验设置的差异。CPA主要设计用于学习适用于未见细胞类型的共享扰动嵌入,这与我们专注于完全未见扰动的目标有所偏离。为确保公平的基准测试,我们将CPA从最终比较中排除。
我们在相同的设置下训练了所有模型并报告评估结果。首先,使用所有基因的总计数对每个细胞的基因表达值进行标准化,并应用对数转换。随后,我们选择了5,000个高变异基因,并将任何最初未考虑的受扰动基因纳入基因集。在实验中,对于所有三个数据集中的单基因扰动,对扰动进行拆分以确保测试扰动在训练中未见过,即训练集中的细胞未经历过任何测试扰动。对于Norman等人数据集中的双基因扰动,训练-测试分割包括三个难度递增的场景:
-
训练集中两个基因都见过;
-
训练集中一个基因未见过;
-
训练集中两个基因都未见过。
为评估扰动预测的准确性,我们检查了扰动后和对照细胞状态之间的表达变化("delta")。我们计算了预测和观察到的数据表达变化之间的皮尔逊相关性,表示为Pearson delta。我们还报告了这些皮尔逊指标在表达差异最大的前20个基因上的结果。因此,我们为差异表达条件提供了额外的评估指标,即差异表达基因上的Pearson delta。此外,我们测试了预测基因表达与真实表达值之间的皮尔逊相关性(Pearson)。在补充说明9中进行的比较分析表明,基于delta的评估比仅与真实表达水平的相关性能更真实地反映模型性能。
对于基于聚类的生物学验证,我们首先从scGPT模型中获取了每个扰动条件的代表性基因表达谱。scGPT从单个样本对照基因表达向量(即大小为1×M基因)预测每个扰动条件的代表性扰动响应,该向量是通过对数据集中所有对照细胞的基因表达取平均值获得的。Norman等人的数据集包含105个独特的受扰动基因,这产生了总共5,565个独特的扰动组合需要预测。
我们将高维预测扰动响应投影到二维UMAP上。我们首先将UMAP图与Norman等人原始论文中发现的功能组进行了比较,其中236个扰动实验是基于真实扰动响应进行聚类,并通过标记基因表达注释其功能角色。我们检查了scGPT预测的UMAP投影与原始论文中发现的功能分组之间的一致性。然后,我们分析了scGPT预测的UMAP中存在的亚聚类。使用0.5的Leiden聚类分辨率,在预测扰动响应的UMAP中识别出54个亚聚类。我们用最频繁出现的受扰动基因作为其主导基因来注释每个聚类。
对于反向扰动预测任务,我们从Norman数据集中选择了20个基因来构建扰动用例,并微调和测试新的扰动模型。这个20个基因的子集是通过最大化基于scGPT训练-测试分割的训练和测试案例的真实扰动数据比例来选择的。该选定子空间在210个独特扰动组合中包含39个训练案例、3个验证案例和7个测试案例。其余为无实验结果的未见案例。
反向扰动预测遵循top-K检索任务设置:我们使用所有210个扰动条件的预测响应作为参考数据,使用七个测试案例的真实响应作为查询集。目标是检索产生最相似响应的顶部扰动条件。对于参考数据,我们从30个随机采样的对照细胞获得预测响应,以增加多样性,而不是每个扰动条件只有一个代表性基因表达谱。这产生了包含6,300个预测后扰动基因表达谱的参考数据库。对于每个X+Y扰动的测试案例,我们使用所有经历过X+Y扰动的细胞的真实基因表达谱作为查询集。
对于top-K检索,我们设计了一个包含两轮选择的集成投票策略。在第一轮中,每个单独的查询细胞通过欧氏距离从参考数据集中选择其最相似的K个表达谱。在第二轮中,我们根据所有查询细胞的投票数对候选扰动条件进行排名。我们报告第二轮集成投票后得票最多的K个扰动条件作为预测的源扰动条件。
我们通过修改后的top-K准确率指标评估检索性能,包括正确检索(即精确匹配)和相关检索(即与实际扰动组合中至少匹配一个基因)。我们报告了scGPT在五次不同随机种子运行的平均分数。我们将scGPT的检索性能与GEARS和差异基因进行了基准对比。对于与GEARS的基准对比,我们使用GEARS预测的表达谱作为参考数据库,并报告相同的top-K检索指标。对于差异基因,我们通过Wilcoxon秩和检验在受扰动细胞和对照细胞之间识别每个测试扰动的前两个差异表达基因。我们将这两个差异表达最显著的基因视为预测的top-1双基因扰动组合。我们仅报告差异表达的top-1检索指标,因为随着差异表达基因列表的扩大,扰动组合变得模糊不清。
单细胞RNA测序批次整合
在这项工作中,我们将scGPT的性能与其他三种方法进行了比较,即Seurat、Harmony和scVI。评估涵盖了三个整合数据集的批次校正和细胞类型聚类:COVID-19、PBMC 10k和perirhinal cortex(外周皮层)。Harmony和scVI在最近的整合基准测试中被强调为表现最佳的方法。为确保公平比较,所有方法都使用相同数量的1,200个高变异基因(HVGs)作为输入。通过考虑所有基因的总计数对每个细胞的基因表达值进行标准化,随后进行对数转换。训练完成后获得集成的细胞嵌入,用于评估。
对集成的细胞嵌入的评估是使用生物学保守性指标进行的。这些指标包括NMIcell、ARIcell和ASWcell。这些分数用于衡量派生的细胞类型聚类与真实标签之间的一致性。为便于比较,我们还计算了这些指标的平均值,称为AvgBIO。
此外,我们报告了批次校正指标来评估批次混合效果。批次校正性能通过批次聚类的平均轮廓宽度的倒数(表示为ASWbatch)和图连通性度量(表示为GraphConn)来量化。我们计算ASWbatch和GraphConn的平均值作为AvgBATCH,以总结批次混合性能。此外,我们引入了一个总体得分,它是AvgBIO和AvgBATCH的加权和,这与最近的基准研究中采用的方法一致。
单细胞多组学整合
我们在两种整合设置(配对和镶嵌)中对scGPT进行了基准测试,与最近的单细胞多组学整合方法Seurat (v.4)、scGLUE和scMoMat进行比较。在配对数据整合实验中,我们首先使用10x Multiome PBMC数据集(包含RNA和ATAC-seq数据)对scGPT与scGLUE和Seurat (v.4)进行了基准比较。所有方法都使用相同的1,200个高变异基因和4,000个高变异峰值作为输入。
我们进一步在BMMC数据集(包含配对的RNA和蛋白质数据)上将scGPT与Seurat (v.4)进行了基准比较。在这种情况下,我们没有对scGLUE进行基准测试,因为该方法并非专门设计用于建模蛋白质数据,这是为了公平比较。同样,使用相同的1,200个高变异基因和所有134个蛋白质作为输入。
在镶嵌数据整合实验中,我们在ASAP PBMC数据集上将scGPT与scMoMat进行了基准比较。总共使用1,200个高变异基因、4,000个高变异峰值和所有216个蛋白质特征作为两种方法的输入。在保持输入特征集一致的同时,我们使用每种方法的自定义预处理流程来标准化表达值。训练完成后获取集成的细胞嵌入用于评估。
在配对和镶嵌数据整合设置的所有三个数据集中,我们使用四个生物学保守性指标来评估细胞嵌入质量:NMIcell、ARIcell、ASWcell和AvgBIO。由于在这三个数据集中,BMMC(配对)和ASAP PBMC(镶嵌)包含多个批次,我们进一步使用三个批次校正指标评估了不同组学批次的混合情况:ASWbatch、GraphConn和AvgBATCH。在镶嵌整合实验中还报告了一个总体得分。
基因调控网络推断
我们对scGPT基因嵌入相似性网络进行了验证,将其与已知的HLA和CD基因网络进行比较。
对于每个网络,我们首先通过筛选具有特定前缀(即HLA-和CD-)的基因名称来定义相关基因集。然后,我们筛选了来自Reactome 2022数据库中免疫系统R-HSA-168256通路中涉及的基因。对于CD基因,我们使用了来自免疫人类数据集的高变异基因的共同基因集,以便于比较预训练和微调模型。
我们从scGPT模型中提取这些选定基因的基因嵌入,并构建了一个k近邻相似性网络。通过选择余弦相似度大于特定阈值的边(即HLA网络为0.5,CD基因网络为0.4),我们突出显示了强连接的子网络。然后,我们将这些子网络与免疫系统中已知的功能群进行了比较。
此外,我们通过通路富集分析验证了由scGPT模型提取的基因程序的质量。我们使用每个基因程序作为输入基因列表,并选择具有统计显著性的通路作为"通路命中"。P值阈值经过Bonferroni校正后设定为0.05(https://mathworld.wolfram.com/BonferroniCorrection.html),校正基于总测试次数,即基因程序数量乘以通路测试数量。我们报告了在Reactome 2022数据库(https://reactome.org/)中的通路命中数。
作为基准,我们将结果与从基线共表达图中提取的基因程序进行了比较。共表达图是通过免疫人类数据集中基因标准化表达的Pearson相关性定义的。为确保与scGPT网络具有相似的模块性,我们将该图稀疏化为k近邻相似性网络(k=15)。按照与scGPT相同的流程,我们通过Leiden聚类从基因集群中识别基因程序。
作为敏感性分析,我们报告了scGPT和共表达方法在不同Leiden分辨率(1、5、10、20、30、40、50和60)下的通路命中情况。我们进一步在Leiden分辨率40下检查了每种方法识别的共同和独特通路,以深入了解性能差异。
我们在ChIP-Atlas数据库中验证了基于scGPT注意力的最受影响基因选择方法,该数据库包含已知转录因子的实验验证基因靶点。我们首先通过将Adamson扰动数据集中的扰动基因列表与ChIP-Atlas交叉检查,选择了两个示例转录因子,它们由DDIT3和BHLHE40编码。对于每个转录因子,我们通过将"差异"设置下注意力选择的前20个最受影响基因与已验证的基因靶点进行比较来进行验证。需要注意的是,在"差异"设置下,前20个基因是基于扰动后变化通过检查扰动注意力图和对照注意力图之间的差异来选择的。
真实的基因靶点列表是通过从ChIP-Atlas中筛选人类基因(hg38)获得的,这些基因的转录起始位点位于转录因子峰值调用区间的10kb距离内。我们报告了注意力选择的前20个基因靶点与真实靶点基因之间的重叠数量。
随后,我们比较了三种最受影响基因选择方法(即对照组、扰动组和差异组)中前100个选定基因之间的重叠情况。这三组前100个基因集合的重叠和差异通过Venn图进行可视化。我们还在Reactome数据库中进一步验证了这些顶部基因与转录因子一起参与的通路。通路命中和基因重叠百分比通过热图进行可视化。
我们进一步验证了功能相关转录因子组的最受影响基因选择分析。我们使用了由Replogle等人确定的两个示例扰动组,这些组分别与mRNA多聚腺苷化和组蛋白乙酰化相关。通过与ChIP-Atlas数据库交叉检验,我们从扰动基因列表中选择了转录因子。因此,我们得到了以下具有功能注释的转录因子基因组:
-
用于
mRNA多聚腺苷化的CPSF2、CPSF3、CPSF4和CSTF3; -
用于组蛋白乙酰化的
KAT8、MCRS1和YEATS4。
需要注意的是,KANSL3和CPSF1也被Replogle等人列入这两个功能组。然而,由于这些基因对应的转录因子在用于构建注意力图的1,200个HVG中有超过十个注释的基因靶点,因此在后续分析中将其排除。
按照之前对单个转录因子的分析方法,我们首先使用"扰动"设置为每个转录因子选择前20个最受影响的基因,并在ChIP-Atlas数据库中验证基因靶点。对于每个功能组,我们随后报告了所有转录因子(连同转录因子本身)的前100个最受影响基因富集的Reactome通路。具体而言,对于mRNA多聚腺苷化,我们从CPSF2、CPSF3、CPSF4和CSTF3的前25个最受影响基因的并集中获得前100个最受影响基因;对于组蛋白乙酰化,则从KAT8、MCRS和YEATS4的前33个最受影响基因的并集中获得。
为了验证,我们将富集的Reactome通路与Replogle等人的功能注释特定术语进行比较。我们还通过文献检索进一步验证了相关通路。如果满足以下两个标准之一,则认为某个术语是相关的:
-
它包含一个或多个转录因子;
-
其与功能注释的关联得到现有文献支持。
实现细节
预训练的基础模型具有512的嵌入大小,由12个堆叠的转换器块组成,每个块有8个注意力头。全连接层的隐藏层大小为512。在使用3300万个细胞的全人类模型预训练中,我们随机拆分数据,使用99.7%的数据用于训练,0.3%用于验证。对于其他模型(包括器官特异性模型和泛癌症模型)的预训练,我们随机拆分数据,使用97%的数据用于训练,3%用于验证。注意,在预训练中,只有非零表达的基因才会输入到模型中。我们设置最大输入长度为1,200。对于非零基因数量大于最大输入长度的细胞,会在每次迭代时随机采样1,200个输入基因。我们设置要生成的基因比率从0.25、0.50和0.75三个选项中均匀采样。模型使用Adam优化器进行优化,小批量大小为32,初始学习率为0.0001,每个周期后权重衰减0.9。模型总共训练了6个周期。
对于scRNA-seq批次整合、细胞类型注释和扰动预测任务,我们使用了从预训练模型继承的相同模型层配置。在微调过程中,我们以0.0001的学习率开始,每个周期后衰减到90%。对于整合任务,GEP和GEPC的掩码比率设置为0.4,而ECS中的参数β设置为0.6。当与其他损失函数结合时,ECS被赋予权重10。为了将数据集划分为训练集和验证集,我们使用了9:1的比例。模型训练固定持续了15个周期,在每个周期后,在验证集上评估GEP损失值。报告的结果对应于具有最佳验证分数的模型。
对于多组学整合任务,我们加载了预训练模型的基因嵌入,并对任何新的标记(即基因、ATAC峰值或蛋白质)使用相同的512嵌入大小。主模型设置为具有4个堆叠的转换器块,每个块有8个注意力头和512的隐藏层大小。除了预训练的嵌入权重外,所有层都重新初始化。每个数据集以9:1的比例分为训练集和评估集。我们对批次整合使用了1.0的DAR权重。我们使用0.001的初始学习率,每个周期后权重衰减0.95。我们以16的批量大小训练模型固定25个周期,并同样报告了最佳验证模型。
我们使用PyTorch来实现scGPT神经网络模型。Scanpy Python库用于基因表达预处理,包括标准化、对数转换和HVG选择。我们在染色质可及性数据上使用EpiScanpy Python库进行高变异峰值选择。在scRNA-seq批次整合和scMultiomic整合任务中,使用scib.metrics中的实现计算评估指标。在细胞注释任务中,使用scikit-learn包实现评估指标。在GRN推断任务中,使用Scanpy库进行相似性图构建和Leiden聚类。使用GSEApy包实现通路富集分析。其他依赖项包括torchtext 0.14.0、torch-geometric 2.3.0、flash-attn 1.0.1、pandas 1.3.5、cell-gears 0.0.1、umap-learn 0.5.3、leidenalg 0.8.10和wandb 0.12.3。
数据可用性
所有使用的数据集来源都已在数据集部分中报告。预训练数据集可以从CELLxGENE普查数据库(2023年5月15日发布版本)获取:
对于注释任务,MS数据集从:https://www.ebi.ac.uk/gxa/sc/experiments/E-HCAD-35获取。髓样细胞数据集可以通过GEO数据库访问号GSE154763公开获取。处理后的人类胰腺数据集从:https://github.com/JackieHanLab/TOSICA 获取。
对于参考映射,Lung-Kim数据集可通过曲率癌细胞图谱公开获取:https://www.weizmann.ac.il/sites/3CA/lung。处理后的COVID-19数据集从:https://github.com/theislab/scarches-reproducibility 获取。对于扰动预测任务,Norman和Adamson数据集分别从:
- https://dataverse.harvard.edu/api/access/datafile/6154020
- https://dataverse.harvard.edu/api/access/datafile/6154417
获取。Replogle数据集从:https://gwps.wi.mit.edu/ 获取。
对于批次整合任务,PBMC 10k数据集从scVI tools( https://scvi-tools.org/ )获取,外周皮层数据集从CELLxGENE人类脑细胞图谱1.0版本获取:https://cellxgene.cziscience.com/collections/283d65eb-dd53-496d-adb7-7570c7caa443 。
对于多组学整合任务,10x Multiome PBMC数据集从:https://scglue.readthedocs.io/en/latest/data.html 获取,BMMC数据集可通过GEO数据库访问号GSE194122获取,ASAP PBMC数据集从:https://github.com/PeterZZQ/scMoMaT/tree/main/data/real/ASAP-PBMC 获取。
对于GRN分析,处理后的免疫人类数据集从:https://doi.org/10.6084/m9.figshare.12420968.v8 获取。所有处理后的数据集都可以从:
获取。
代码可用性
scGPT的代码库在GitHub上公开可用( https://github.com/bowang-lab/scGPT ),并在Zenodo存储库( https://doi.org/10.5281/zenodo.10466117 )中提供,采用MIT许可证。
补充信息
补充信息1–12,表1–7和图1–13请在这里查看:41592_2024_2201_MOESM1_ESM.pdf 。
参考文献
-
Silverman, A. D., Karim, A. S. & Jewett, M. C. Cell-free gene expression: an expanded repertoire of applications. Nat. Rev. Genet. 21, 151-170 (2020).
-
Preissl, S., Gaulton, K. J. & Ren, B. Characterizing cis-regulatory elements using single-cell epigenomics. Nat. Rev. Genet. 24, 21-43 (2022).
-
Ding, J., Sharon, N. & Bar-Joseph, Z. Temporal modelling using single-cell transcriptomics. Nat. Rev. Genet. 23, 355-368 (2022).
-
Wagner, D. E. & Klein, A. M. Lineage tracing meets single-cell omics: opportunities and challenges. Nat. Rev. Genet. 21, 410-427 (2020).
-
Regev, A. Science Forum: the Human Cell Atlas. eLife 6, e27041 (2017).
-
Han, X. Mapping the mouse cell atlas by Microwell-seq. Cell 172, 1091-1107 (2018).
-
Angerer, P. et al. Single cells make big data: new challenges and opportunities in transcriptomics. Curr. Opin. Syst. Biol. 4, 85-91 (2017).
-
Subramanian, I., Verma, S., Kumar, S., Jere, A. & Anamika, K. Multi-omics data integration, interpretation, and its application. Bioinform. Biol. Insights 14, 1177932219899051 (2020).
-
Miao, Z., Humphreys, B. D., McMahon, A. P. & Kim, J. Multi-omics integration in the age of million single-cell data. Nat. Rev. Nephrol. 17, 710-724 (2021).
-
Lotfollahi, M., Wolf, F. A. & Theis, F. J. scGen predicts single-cell perturbation responses. Nat. Methods 16, 715-721 (2019).
-
Lotfollahi, M. Predicting cellular responses to complex perturbations in high-throughput screens. Mol. Syst. Biol. 19, e11517 (2023).
-
Lotfollahi, M. Mapping single-cell data to reference atlases by transfer learning. Nat. Biotechnol. 40, 121-130 (2022).
-
Cao, Z.-J. & Gao, G. Multi-omics single-cell data integration and regulatory inference with graph-linked embedding. Nat. Biotechnol. 40, 1458-1466 (2022).
-
Zhang, Z. et al. scMoMat jointly performs single cell mosaic integration and multi-modal bio-marker detection. Nat. Commun. 14, 384 (2023).
-
Bommasani, R. et al. On the opportunities and risks of foundation models. Preprint at https://doi.org/10.48550/arXiv.2108.07258 (2021).
-
Moor, M. et al. Foundation models for generalist medical artificial intelligence. Nature 616, 259-265 (2023).
-
Vaswani, A. et al. Attention is all you need. Adv. Neural Inf. Process. Syst. 6000-6010 (NeurIPS, 2017).
-
Ramesh, A., Dhariwal, P., Nichol, A., Chu, C. & Chen, M. Hierarchical text-conditional image generation with CLIP latents. Preprint at https://doi.org/10.48550/arXiv.2204.06125 (2022).
-
Brown, T. Language models are few-shot learners. Adv. Neural. Inf. Process. Syst. 1877-1901 (NeurIPS, 2020).
-
OpenAI team. GPT-4 technical report. Preprint at https://doi.org/10.48550/arXiv.2303.08774 (2023).
-
Avsec, Z. et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 18, 1196-1203 (2021).
-
Gururangan, S. et al. Don’t stop pretraining: adapt language models to domains and tasks. In Proc. 58th Annual Meeting of the Association for Computational Linguistics 8342-8360 (ACL, 2020).
-
Qiu, X. et al. Pre-trained models for natural language processing: a survey. Sci. China Technol. Sci. 63, 1872-1897 (2020).
-
Liu, J., Fan, Z., Zhao, W. & Zhou, X. Machine intelligence in single-cell data analysis: advances and new challenges. Front. Genet. 12, 655536 (2021).
-
Oller-Moreno, S., Kloiber, K., Machart, P. & Bonn, S. Algorithmic advances in machine learning for single-cell expression analysis. Curr. Opin. Syst. Biol. 25, 27-33 (2021).
-
Ji, Y., Lotfollahi, M., Wolf, F. A. & Theis, F. J. Machine learning for perturbational single-cell omics. Cell Syst. 12, 522-537 (2021).
-
Theodoris, C. V. et al. Transfer learning enables prediction in network biology. Nature 618, 616-624 (2023).
-
McInnes, L., Healy, J. & Melville, J. UMAP: uniform manifold approximation and projection for dimension reduction. Preprint at https://doi.org/10.48550/arXiv.1802.03426 (2018).
-
Schirmer, L. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 573, 75-82 (2019).
-
Cheng, S. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell 184, 792-809 (2021).
-
Chen, J. et al. Transformer for one stop interpretable cell type annotation. Nat. Commun. 14, 223 (2023).
-
Yang, F. et al. scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data. Nat. Mach. Intell. 4, 852-866 (2022).
-
Adamson, B. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867-1882 (2016).
-
Replogle, J. M. Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell 185, 2559-2575 (2022).
-
Norman, T. M. et al. Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science 365, 786-793 (2019).
-
Roohani, Y., Huang, K. & Leskovec, J. Predicting transcriptional outcomes of novel multigene perturbations with GEARS. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-01905-6 (2023).
-
Traag, V. A., Waltman, L. & Van Eck, N. J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019).
-
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053-1058 (2018).
-
Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495-502 (2015).
-
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289-1296 (2019).
-
Gayoso, A. A Python library for probabilistic analysis of single-cell omics data. Nat. Biotechnol. 40, 163-166 (2022).
-
Siletti, K. Transcriptomic diversity of cell types across the adult human brain. Science 382, eadd7046 (2023).
-
PBMC from a healthy donor, single cell multiome ATAC gene expression demonstration data by Cell Ranger ARC 1.0.0. 10X Genomics https://support.10xgenomics.com/single-cell-multiome-atac-gex/datasets/1.0.0/pbmc_granulocyte_sorted_10k (2020).
-
Hao, Y. Integrated analysis of multimodal single-cell data. Cell 184, 3573-3587 (2021).
-
Luecken, M. et al. A sandbox for prediction and integration of DNA, RNA, and proteins in single cells. In Proceedings of the Neural Information Processing Systems Track on Datasets and Benchmarks 13 (NeurIPS, 2021).
-
Mimitou, E. P. Scalable, multimodal profiling of chromatin accessibility, gene expression and protein levels in single cells. Nat. Biotechnol. 39, 1246-1258 (2021).
-
Pratapa, A., Jalihal, A. P., Law, J. N., Bharadwaj, A. & Murali, T. M. Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data. Nat. Methods 17, 147-154 (2020).
-
Choo, S. Y. The HLA system: genetics, immunology, clinical testing, and clinical implications. Yonsei Med. J. 48, 11-23 (2007).
-
Norman, P. S. Immunobiology: the immune system in health and disease. J. Allergy Clin. Immunol. 96, 274 (1995).
-
Luecken, M. D. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 19, 41-50 (2022).
-
Zou, Z., Ohta, T., Miura, F. & Oki, S. ChIP-Atlas 2021 update: a data-mining suite for exploring epigenomic landscapes by fully integrating ChIP-seq, ATAC-seq and Bisulfite-seq data. Nucleic Acids Res. 50, W175-W182 (2022).
-
Yang, H., Niemeijer, M., van de Water, B. & Beltman, J. B. ATF6 is a critical determinant of CHOP dynamics during the unfolded protein response. iScience 23, 100860 (2020).
-
Yoshida, H. et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 20, 6755-6767 (2000).
-
Kaplan, J. et al. Scaling laws for neural language models. Preprint at https://doi.org/10.48550/arXiv.2001.08361 (2020).
-
Haque, A., Engel, J., Teichmann, S. A. & Lönnberg, T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 9, 1-12 (2017).
-
Devlin, J., Chang, M. W., Lee, K. & Toutanova, K. BERT: pre-training of deep bidirectional transformers for language understanding. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics 4171-4186 (ACL, 2019).
-
Dao, T., Fu, D., Ermon, S., Rudra, A. & Ré, C. FlashAttention: fast and memory-efficient exact attention with IO-Awareness. Adv. Neural. Inf. Process. Syst. 16344-16359 (NeurIPS, 2022).
-
Wang, S., Li, B. Z., Khabsa, M., Fang, H. & Ma, H. Linformer: self-attention with linear complexity. Preprint at https://doi.org/10.48550/arXiv.2006.04768 (2020).
-
Katharopoulos, A., Vyas, A., Pappas, N. & Fleuret, F. Transformers are RNNs: fast autoregressive transformers with linear attention. In Proc. 37th International Conference on Machine Learning 5156-5165 (PMLR, 2020).
-
Liu, Y. RoBERTa: a robustly optimized BERT pretraining approach. Preprint at https://doi.org/10.48550/arXiv.1907.11692 (2019).
-
Bubeck, S. et al. Sparks of artificial general intelligence: early experiments with GPT-4. Preprint at https://doi.org/10.48550/arXiv.2303.12712 (2023).
-
Liu, C. et al. Guided similarity separation for image retrieval. Adv. Neural. Inf. Process. Syst. 1556-1566 (NeurIPS, 2019).
-
Eisenstein, M. Single-cell RNA-seq analysis software providers scramble to offer solutions. Nat. Biotechnol. 38, 254-257 (2020).
-
Tran, H. T. N. et al. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 21, 12 (2020).
-
Ganin, Y. & Lempitsky, V. Unsupervised domain adaptation by backpropagation. In Proc. 32nd International Conference on Machine Learning 1180-1189 (PMLR, 2015).
-
Cecilia, N. Identification of transcriptional programs using dense vector representations defined by mutual information with GeneVector. Nat. Commun. 14, 4400 (2023).
-
Kim, N. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 11, 2285 (2020).
-
Paszke, A. PyTorch: an imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Sys. 1-12 (NeurIPS, 2019).
-
Wolf, F. A., Angerer, P. & Theis, F. J. Scanpy: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
-
Danese, A. et al. EpiScanpy: integrated single-cell epigenomic analysis. Nat. Commun. 12, 5228 (2021).
-
Fang, Z., Liu, X. & Peltz, G. GSEApy: a comprehensive package for performing gene set enrichment analysis in Python. Bioinformatics 39, btac757 (2023).
-
Wang, C. Processed datasets used in the scGPT foundation model. Figshare https://doi.org/10.6084/m9.figshare.24954519.v1 (2024).
-
Cui, H., Wang, C. & Pang, K. Codebase for scGPT: towards building a foundation model for single-cell multi-omics using generative AI. Zenodo https://doi.org/10.5281/zenodo.10466117 (2024).
